Синдром Корнелии де Ланге
| Синдром Корнелии де Ланге | |
|---|---|
 | |
| МКБ-10 | Q87.1 |
| МКБ-10-КМ | Q87.1 |
| МКБ-9 | 759.89 |
| OMIM | 122470 |
| DiseasesDB | 29651 |
| MeSH | D003635 |
Синдром Корнелии де Ланге (синдром Брахмана-Ланге) — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000.
Впервые случай такого заболевания описан в 1916 году немецким врачом В. Брахманом (W. R. C. Brachmann), название синдром получил по имени голландского педиатра Корнелии де Ланге (Cornelia de Lange), в 1933 году описавшей синдром на основе анализа пяти случаев заболевания.
Генетика
Синдром Корнелии де Ланге относится к доминантно-наследуемым заболеваниям, большинство случаев являются спорадическими, возникшими de novo. Синдром является генетически гетерогенным. Примерно половина случаев обусловлена мутациями в гене NIPBL, около 5 % случаев — мутациями в гене SMC1A, кодирующем субъединицу белкового комплекса когезина. Один описанный случай связан с мутацией в гене SMC3, который также кодирует одну из субъединиц когезина.
В ряде случаев при цитогенетическом исследовании находят микродупликацию локусов q25 — q29 хромосомы 3.
Клиническая картина
- Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
- Брахицефалия (укорочение черепа в сагиттальном направлении, в результате чего поперечный размер головы увеличивается, а продольный уменьшается);
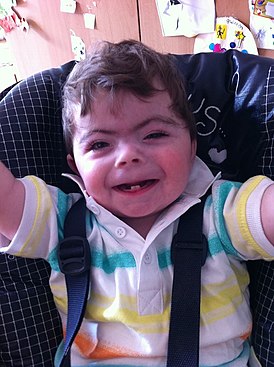
- Тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа;
- Микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов;
- Миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва;
- Маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев;
- Мраморная кожа;
- Гипоплазия сосков;
- Гипертрихоз;
- Судороги;
- Врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм) и др.
У части больных наблюдается бег по кругу, стереотипные движения руками и самоповреждения.
У всех больных отмечаются отставание в росте, в 80 % случаев — тяжёлая умственная отсталость (имбецильность), в оставшихся 20 % менее выраженные интеллектуальные нарушения; типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй — с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития.