Острый инфаркт миокарда
Инфаркт миокарда (ИМ) — это некроз сердечной мышцы, развивающийся в результате длительной ишемии. Учитывая постоянные воздействия на коронарные артерии вследствие их растяжения и сдавления работающим миокардом, широкий диапазон потребности миокарда в кислороде, кажется удивительным, что тяжелая ишемия миокарда не встречается еще чаще. Однако механизмы саморегуляции коронарных артерий, описанные выше, обычно обеспечивают адекватную доставку кислорода к миокарду даже при наличии атеросклеротических бляшек. Однако при расстройстве обычных защитных механизмов может развиться длительная ишемия или инфаркт миокарда.
Приблизительно у 1,5 млн граждан США ежегодно развивается ИМ, и примерно у одной трети пациентов он заканчивается фатально. Несмотря на эту драматическую статистику, в течение последних трех десятилетий отмечалось постоянное снижение смертности от ИМ, что было вызвано как лучшим пониманием патофизиологии болезни, так и быстрым развитием новых методов лечения.
Как и при других проявлениях коронарной болезни сердца, осложнения ИМ часто связаны с продолжительностью и выраженностью дисбаланса между потребностью миокарда в кислороде и его доставкой, а долгосрочный прогноз в основном зависит от массы некротизированного миокарда. В этой главе мы рассмотрим события, которые ведут к инфаркту миокарда, последующие патоморфологические и функциональные изменения и терапевтические воздействия, которые могут противодействовать этим патофизиологическим механизмам.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Приблизительно 90% инфарктов миокарда развиваются вследствие образования тромба, перекрывающего просвет атеросклеротически измененной коронарной артерии. Сужение просвета бляшкой за счет тромба превращается в полную окклюзию. Тромбообразование происходит вследствие взаимодействия между атеросклеротической бляшкой и коронарным эндотелием, циркулирующими тромбоцитами и динамическим тонусом сосудистой стенки; эти факторы, как мы увидим далее, перевешивают естественные защитные механизмы.
Система гемостаза в норме
При повреждении нормального кровеносного сосуда нарушается целостность поверхности эндотелия и тромбогенная соединительная ткань входит в соприкосновение с кровотоком. Первичный гемостаз является первой линией защиты от кровотечения. Этот процесс начинается в течение нескольких секунд после повреждения сосуда и осуществляется циркулирующими тромбоцитами, которые прилипают к коллагену субэндотелиального слоя и агрегируются, формируя «тромбоцитарную пробку». Во время формирования первичного тромбоцитарного тромба под действием субэндотелиального тканевого фактора активируется свертывающая система плазмы; так начинается процесс вторичного гемостаза. Белки свертывающей системы, вовлеченные во вторичный гемостаз, последовательно активируются на месте повреждения, где, в конце концов, под действием тромбина образуется фибриновый сгусток. Образующийся тромб стабилизирует и укрепляет «тромбоцитарную пробку».
В норме система гемостаза минимализирует кровопотерю при повреждении сосуда, но этот физиологический ответ мало чем отличается от патологического процесса коронарного тромбоза, запускаемого разрывом атеросклеротических бляшек.
Эндогенные антитромботические механизмы
Нормальные кровеносные сосуды, включая коронарные, имеют собственные защитные механизмы, которые предупреждают спонтанный тромбоз и окклюзию, некоторые из них показаны на рисунке 7.1.
ИНАКТИВАЦИЯ СВЕРТЫВАЮЩИХ ФАКТОРОВ
Несколько естественных ингибиторов регулируют процесс гемокоагуляции, противодействуя тромбообразованию и поддерживая кровоток. Наиболее важными из них являются антитромбин III, белки С и S и ингибитор тканевого фактора.
Антитромбин (AT III) — это белок плазмы крови, необратимо связывающийся с тромбином и другими факторами системы свертывания, инактивируя их и способствуя их удалению из кровотока (механизм 1 на рис. 7.1). Эффективность AT III тысячекратно увеличивается при связывании с гепаран сульфатом, гепариноподобной молекулой, имеющейся в норме на обращенной в просвет поверхности эндотелия.
Белок С/Белок S/Тромбомодулин представляют естественную противосвертывающую систему, которая инактивирует факторы «акцелерации» системы гемостаза (то есть, факторы Va и Villa). Белок С синтезируется в печени и циркулирует в неактивном состоянии. Тромбомодулин является тромбинсвязывающим рецептором, в норме представленным на поверхности эндотелия. Связанный с тромбомодулином тромбин не может превращать фибриноген в фибрин (конечная реакция тромбообразования). Вместо этого комплекс тромбин-тромбомодулин активирует белок С. Активированный белок С способствует деградации факторов Va и Villa (механизм 2 на рис. 7.1), ингибируя таким образом гемокоагуляцию. Наличе циркулирующего белка S усиливает ингибиторную функцию белка С.
Ингибитор тканевого фактора (ИТФ) является недавно открытым ингибитором сериновой протеиназы плазмы, активируемой фактором свертывания Ха. Комплекс: фактор Ха/ИТФ связывается с комплексом: тканевой фактор/фактор Vila, запускающий в норме «внешний» путь свертывания крови, и инактивирует его (механизм 3 на рис. 7.1). Таким образом, ИТФ подавляет гемокоагуляцию по механизму отрицательной обратной связи.
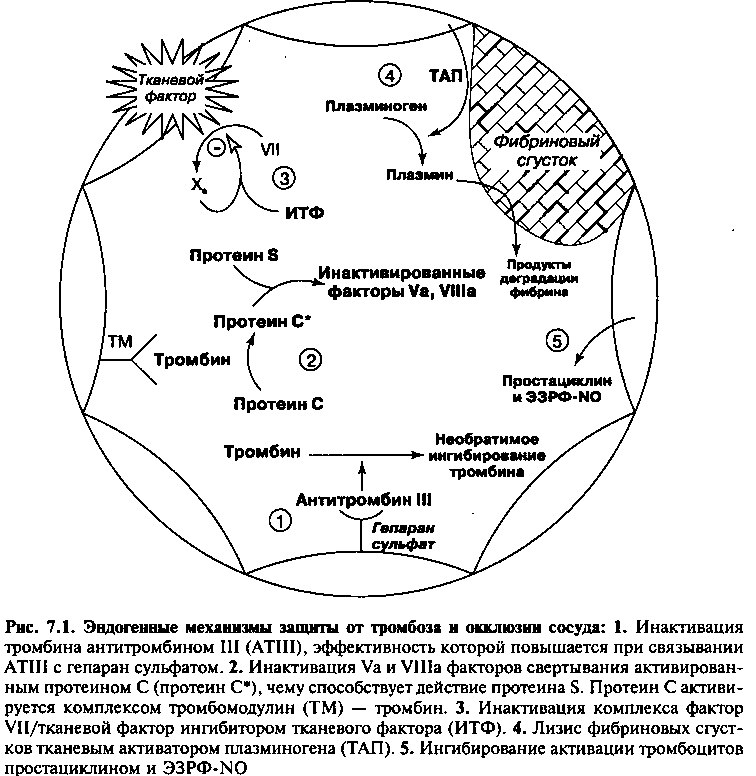
ЛИЗИС ФИБРИНОВЫХ СГУСТКОВ
Тканевой активатор плазминогена (ТАП) — это белок, секретируемый эндотелиальными клетками в ответ на многочисленные пусковые факторы тромбообразования. ТАП превращает белок плазминоген в активную форму — плазмин, который в свою очередь является ферментом, катализирующим деградацию фибриновых тромбов (механизм 4 на рис. 7.1). Когда ТАП связывается с фибрином в формирующемся тромбе, его способность превращать плазминоген в плазмин возрастает в сотни раз.
ЭНДОГЕННОЕ ИНГИБИРОВАНИЕ АГРЕГАЦИИ ТРОМБОЦИТОВ И ВАЗОДИЛА ТАЦИЯ
Простациклин синтезируется и секретируется клетками эндотелия, как описано в главе 6. Он увеличивает уровень циклического АМФ внутри тромбоцита и таким образом сильно подавляет активацию и агрегацию тромбоцитов (механизм 5 на рис. 7.1). Простациклин также подавляет гемокоагуляцию непрямым образом посредством своего мощного вазодилатирующего действия. Вазодилатация предохраняет от тромбоза, усиливая кровоток (и предотвращая таким образом контакт между прокоагулянтами) и уменьшая напряжение сдвига (один из факторов активации тромбоцитов).
Эндотелий-зависимый фактор релаксации/оксид азота (ЭЗФР-NO) также секретируется клетками эндотелия, как это показано в главе 6. Он локально ингибирует активацию тромбоцитов (механизм 5 на рис. 7.1) и действует также как мощный вазодилататор.
Патогенез коронарного тромбоза
В норме вышеописанные механизмы противодействуют спонтанному образованию тромба внутри сосуда. Однако ассоциированные с атеросклерозом отклонения могут перевесить эти защитные механизмы и привести к коронаротромбозу и окклюзии сосуда (рис. 7.2). Атеросклероз способствует формированию тромба посредством: 1) разрыва бляшки, в результате чего тромбогенные вещества входят в соприкосновение с фор-
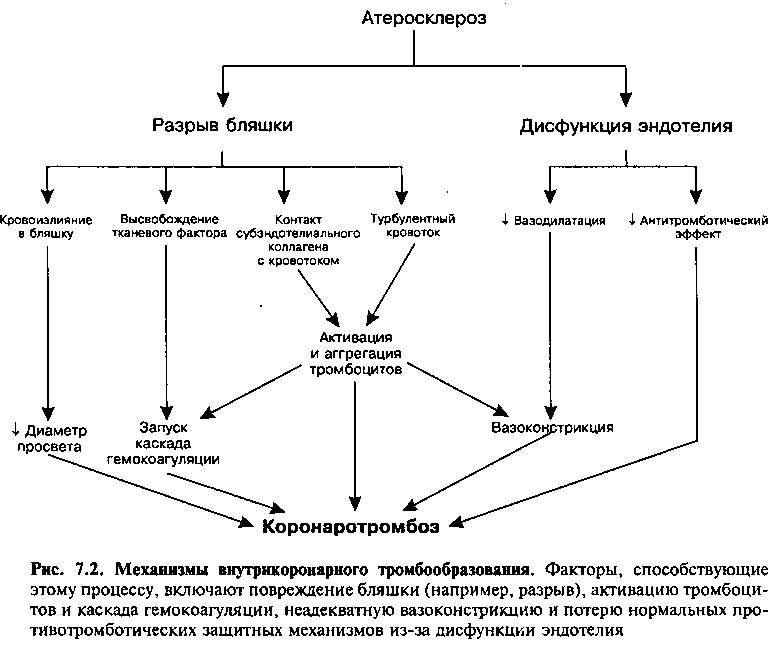
менными элементами крови и 2) дисфункции эндотелия с потерей нормальных защитных антитромботических и вазодилатирующих свойств.
Разрыв атеросклеротической бляшки считается основным пусковым фактором коронаротромбоза. Причины, приводящие к разрыву бляшки, видимо, являются многофакторными и включают: 1) химические факторы, увеличивающие вероятность разрыва бляшки, и 2) физические повреждения бляшек. Бляшки имеют внешнюю фиброзную покрышку, которая окружает некротическое ядро, нагруженное липидами. Доказано, что вещества, высвобождаемые из лейкоцитов внутрь бляшки, могут нарушать целостность фиброзной покрышки. Например, Т-лимфоциты вырабатывают гамма-интерферон, который ингибирует синтез коллагена гладкомышечными клетками и, таким образом, уменьшает прочность фиброзной покрышки. Кроме того, клетки внутри атеросклеротических бляшек продуцируют ферменты (например, коллагеназу и желатиназу), которые способствуют деградации интерстициального матрикса, дестабилизируя далее структуру бляшки. Ослабленная, с тонкой покрышкой бляшка подвержена разрывам (особенно в зоне «плеча», то есть ее границы с нормальной стенкой артерии, испытывающей особое напряжение), как спонтанным, так и под действием физических факторов, таких как давление крови в сосуде и воздействие со стороны работающего миокарда.
Инфаркты иногда развиваются под действием определенных триггерных факторов, таких как чрезмерная физическая нагрузка или психоэмоциональный стресс. Активация симпатической нервной системы в этих ситуациях повышает артериальное давление, частоту сердечного ритма и силу желудочковых сокращений, а все эти факторы могут повреждать атеросклеротическую бляшку, вызывая ее трещины и разрывы. Помимо этого, инфаркты наиболее часто развиваются в ранние утренние часы. Вероятно, это связано с тем, что уровень ключевых физиологических стрессорных факторов (систолического артериального давления, вязкости крови и адреналина в плазме) наиболее высок в это время суток, способствуя разрыву уязвимых бляшек.
Как только происходит разрыв бляшки, контакт с субэндотелиальным коллагеном активирует тромбоциты, и тканевый фактор запускает каскад свертывания крови; все вместе взятое ведет к образованию тромба (рис. 7.2). Кровоизлияние в бляшку (см. главу 5), которое также может возникнуть во время разрыва бляшки, может еще больше увеличить ее размер и дополнительно сузить просвет коронарной артерии.
Фиброзная бляшка с фиксированным к ней неокклюзирующим тромбом сужает просвет сосуда и может таким образом вызывать турбулентный ток крови через артерию. Последний подвергает циркулирующие тромбоциты значительному напряжению сдвига и таким образом активирует их, способствуя их агрегации. Далее, внезапные изменения геометрии бляшки (например, кровоизлияние в бляшку) могут дополнительно сужать просвет и усиливать напряжение сдвига и активацию тромбоцитов.
Из активированных тромбоцитов высвобождается содержимое их гранул, которое включает активаторы агрегации тромбоцитов (например, АДФ, фибриноген), активаторы каскада гемокоагуляции (например, фактор Va) и вазоконстрикторы (например, тромбоксан и серотонин). За счет вклада агрегации тромбоцитов в формирующийся тромб просвет сосуда суживается еще больше.
В процессе тромбообразования тромбоцитарные факторы (например, тромбоксан и серотонин), а также содержащийся внутри формирующегося сгустка тромбин стимулируют вазоконстрикцию. В норме сосуды отвечают на тромбоцитарные стимулы вазодилатацией, так как эти вещества способствуют высвобождению эндотелием ЭЗФР-NO и простациклина, влияния которых преобладают над прямым вазоконстрикторным эффектом (см. рис. 6.4). Однако в условиях атеросклероза и снижения продукции эндотелиальных вазодилататоров вазоконстрикция бесконтрольно прогрессирует. Подобным образом содержащийся внутри формирующегося сгустка тромбин при дисфункции эндотелия вызывает выраженный спазм гладкомышечных клеток. Вазоконстрикция вызывает скручивание сосуда, которое может усугублять разрыв бляшки или вызывать транзи-торную окклюзию стенозированного сосуда за счет повышения тонуса артерии. Снижение коронарного кровотока, вызванное вазоконстрикцией, замедляет вымывание белков свертывающей системы, что также повышает тромбогенность.
Дисфункция эвдотелия, которая отмечается даже при умеренном атеросклеротическом поражении коронарных артерий, также повышает вероятность тромбообразования. При дисфункции клетки эндотелия продуцируют сниженные количества вазодилататоров (то есть ЭЗФР-NO и простациклина) и, таким образом, преобладает относительная вазоконстрикция (рис. 7.2). Более того, ЭЗФР-NO и простациклин являются естественными ингибиторами активации тромбоцитов. Таким образом, при их сниженной продукции в условиях дисфункции эндотелия теряется важная защита от тромбообразования.
ПОСЛЕДСТВИЯ КОРОНАРНОГО ТРОМБОЗА
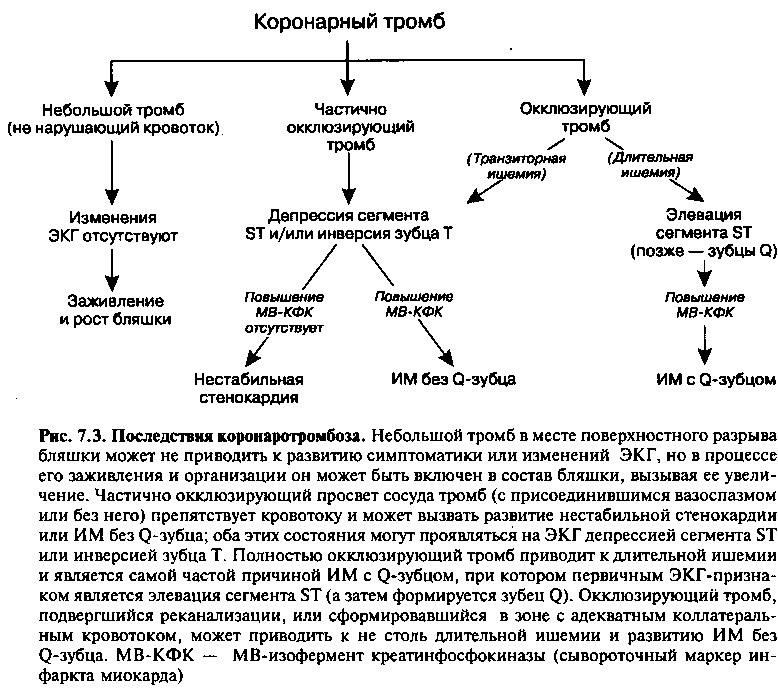
Развитие внутрикоронарного тромбоза приводит к одному из нескольких возможных результатов (рис. 7.3). Например, иногда надрыв бляшки остается небольшим, поверхностным и не прогрессирует, так что формирующийся тромб тоже остается маленьким и не перекрывает просвет сосуда. В этом случае в результате организации тромба он может просто оказаться включенным в состав бляшки или подвергнуться лизису при преобладании естественных антитромботических механизмов. Повторные бессимптомные разрывы бляшек этого типа могут вызывать постепенное прогрессирование стеноза артерии.
Однако более глубокий разрыв бляшки приводит к более массивному ’ контакту с субэндотелиальным коллагеном и тканевым фактором, в результате чего формирующийся крупный тромб может более существенно сужать просвет сосуда. Такого рода обструкция может вызывать длительную тяжелую ишемию и развитие острого коронарного синдрома, например, нестабильной стенокардии или инфаркта миокарда. Если образующийся в месте разрыва бляшки тромб полностью окклюзирует просвет сосуда, кровоток ниже места обструкции прекращается, и длительная ишемия приводит к развитию инфаркта миокарда (обычно — с Q-зубцом). Напротив, при частичной обструкции просвета (или полной, но транзиторной — в результате спонтанной реканализации или прекращения вазоспазма или окклюзии) тяжесть и продолжительность ишемии будут меньше, и более вероятными клиническими проявлениями будут инфаркт миокарда без Q-зубца или нестабильная стенокардия (рис. 7.3).
Иногда инфаркт миокарда без Q-зубца развивается при длительной полной окклюзии сосуда. В этих случаях, судя по всему, имеется достаточный коллатеральный кровоток, который ограничивает распространение некроза и предотвращает развитие крупноочагового Q-инфаркта.
Острый инфаркт миокарда, не связанный с атеросклерозом коронарных артерий
Иногда ишемию и инфаркт миокарда могут вызывать отличные от острого внутрикоронарного тромбообразования причины (табл. 7.1). Их всегда следует заподозрить при развитии ишемических синдромов у пациентов молодого возраста или без коронарных факторов риска. Например, коронарные сосуды могут закупориваться эмболами, оторвавшимися от искусственных или инфицированных (при инфекционном эндокардите) клапанов сердца; к их окклюзии может привести также воспаление при остром васкулите. Иногда интенсивный транзиторный коронароспазм может снижать доставку кислорода к миокарду в такой степени, что это приводит к инфаркту миокарда.
Таблица 7.1. Причины инфаркта миокарда
Атеросклероз с окклюзирующим тромбозом
Васкулиты ( см. главу 15)
Эмболии коронарных артерий (при эндокардите, протезированных клапанах) Врожденные аномалии коронарных артерий Травма или аневризма коронарной артерии
Тяжелый спазм коронарной артерии (самостоятельный или вследствие приема кокаина)
Повышенная вязкость крови (например, при истинной полицитемии, тромбоцитозах)
Существенное повышение потребности миокарда в кислороде (например, стеноз устья аорты)
Одной из причин острого ИМ является злоупотребление кокаином. Кокаин повышает тонус симпатической нервной системы, блокируя пре-синаптический обратный захват норадреналина и повышая выброс катехоламинов надпочечниками; все это вместе взятое может привести к коронароспазму и снижению доставки кислорода к миокарду. Инфаркты в этом случае развиваются в результате повышения потребности миокарда в кислороде (за счет вызванной кокаином симпатической стимуляции сердца, то есть прироста АД и ЧСС) в условиях сниженной его доставки.
ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ И ФИЗИОЛОГИЯ
Патоморфологически инфаркты миокарда характеризуются по глубине некроза стенки желудочка. Наиболее часто встречаются трансмуральные инфаркты, которые захватывают всю толщу миокарда и развиваются в результате длительной, полной окклюзии эпикардиальной коронарной артерии. Напротив, субэндокардиальные инфаркты избирательно поражают внутренние слои миокарда, наиболее подверженные ишемии, поскольку эта зона испытывает наиболее высокое внутрижелудочковое давление, там слабо выражены коллатерали, а кровоток осуществляется проходящими через сокращающийся миокард артериями.
Инфаркт является не мгновенным событием, а продолжительным процессом усугубляющейся ишемии, которая в конце концов выливается в гибель клеток. Миокард, кровоснабжаемый окклюзированным сосудом, погибает быстро. Прилежащие ткани, частично кровоснабжаемые соседними артериями, некротизируются не сразу. С течением времени, однако, ишемия соседних клеток прогрессирует, поскольку потребность в кислороде остается высокой, а его доставка сниженной. Таким образом, зона некроза может постепенно расширяться. Масса миокарда, в конце концов подвергающегося некрозу, таким образом, зависит от: 1) размера зоны, кровоснабжаемой окклюзированным сосудом, 2) ее потребности в кислороде, 3) адекватности коллатерального кровотока, осуществляемого соседними неокклюзированными артериями, и 4) выраженности ответа ткани на ишемию.
Эффективность терапевтического вмешательства и возможные осложнения зависят от особенностей патофизиологических процессов, развивающихся во время инфаркта миокарда. В целом, эти процессы можно разделить на две стадии: ранние изменения, развивающиеся непосредственно во время инфаркта, и поздние изменения, происходящие в восстановительном периоде.
Ранние изменения
Ранние изменения включают гистологическую эволюцию инфаркта и функциональные изменения вследствие влияния недостатка кислорода на сократимость. Кульминацией этих изменений является коагуляционный некроз миокарда, максимально выраженный на 2—4 сутки (рис. 7.4).
ЦИТОЛОГИЧЕСКИЕ ИЗМЕНЕНИЯ
Падение уровня кислорода в миокарде при острой окклюзии сосуда приводит к быстрому переходу от аэробного обмена к анаэробному, гликолитическому. Поскольку окисление жиров и продуктов гликолиза в митохондриях становится невозможным, синтез макроэргических фосфатов резко снижается, а анаэробный гликолиз ведет к накоплению лактата. Снижение pH обусловливает уменьшение податливости и сократимости миокарда уже через 2 минуты после окклюзии сосуда. При отсутствии лечения через 20 минут развивается необратимое повреждение клеток, проявляющееся набуханием митохондрий, краевым расположением ядерного хроматина, дефектами мембраны и истощением запасов гликогена.
В течение нескольких минут содержание АТФ в клетке падает, так как его продукция в результате гликолиза далеко не обеспечивает потребности в нем. Недостаток АТФ подавляет активность трансмембранной Ка+/К+-АТФазы, в результате чего повышаются уровни внутриклеточного Na+ и внеклеточного К+. Повышение Na+ усугубляет отек клетки. Де-

фекты мембраны и повышение внеклеточного К+ приводят к изменению трансмембранного потенциала, создавая условия для возникновения фатальных аритмий (см. главу 11).
Во время ишемии внутри клетки в результате ряда причин накапливается Са++; к этим причинам относятся: 1) активация Na+/ Са++-ионообмен-ного насоса вследствие повышения концентрации Na+ внутри клетки, 2) просачивание Са++ из саркоплазматического ретикулума в цитозоль и 3) изменения в работе потенциалзависимых Са++-каналов и Са++-АТФазы. Вследствие прогрессирующей деструкции клеточной мембраны Са++, поступающий из экстрацеллюлярного пространства, не может быть удален нормальными энергозависимыми механизмами, что отражает переход от обратимого к необратимому повреждению клеток. Высокая внутриклеточная концентрация ионов Са++ считается единым конечным механизмом, приводящим к деструкции клеток миокарда липазами и протеазами.
Тяжелые дефекты клеточных мембран возникают вследствие недостатка макроэргических фосфатов, потери эндогенных антиоксидантов и продукции свободных радикалов нейтрофилами. Протеолитические ферменты, просачивающиеся из некротизированных миоцитов, повреждают соседний миокард; некоторые макромолекулы, попадающие в кровоток, являются диагностическими маркерами острого инфаркта (см. ниже).
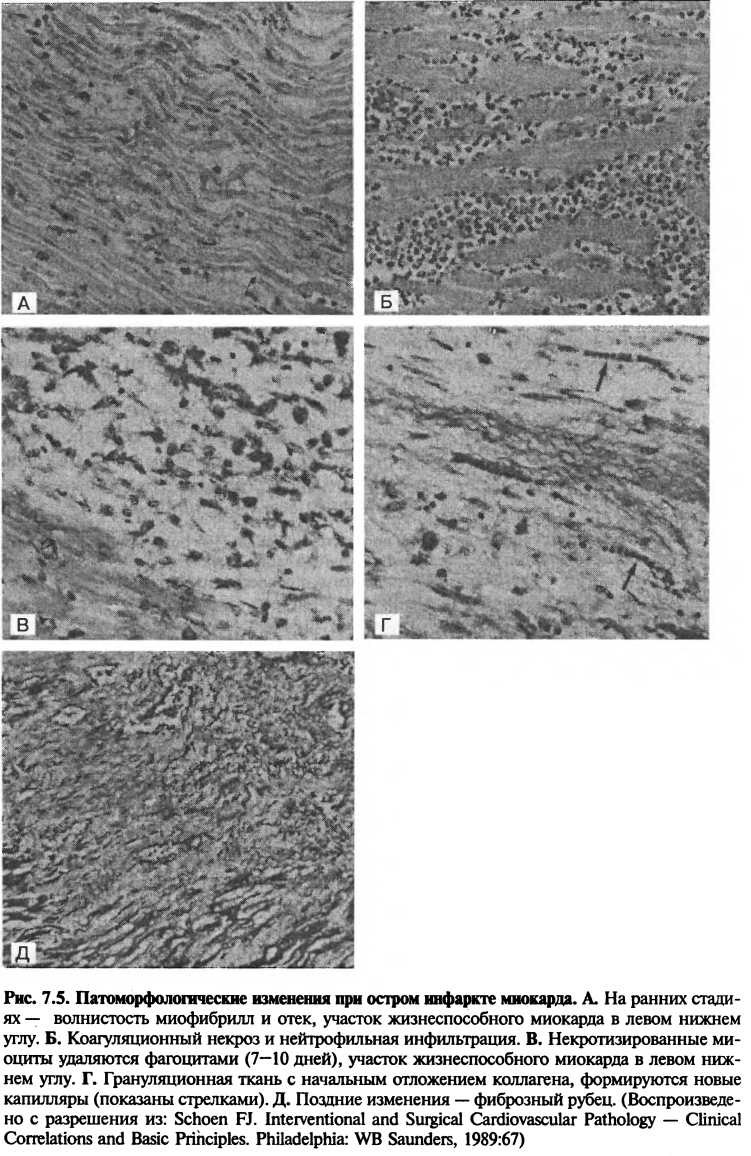
В течение 4—12 часов, по. мере роста проницаемости сосудов под действием медиаторов воспаления и повышения онкотического давления в интерстиции (вследствие просачивания туда внутриклеточных белков), развивается отек миокарда. Наиболее ранним гистологическим изменением, характерными для необратимого повреждения, является волнистость миофибрилл, которая возникает в результате интерстициального отека и «растягивания» в разные стороны кардиомиоцитов под действием сокращений окружающего миокарда (рис. 7.5). По краю инфарктной зоны часто видны пояски сокращения: сократившиеся и консолидированные саркомеры выглядят как яркие эозинофильные полоски.
Приблизительно через 4 часа начинается воспалительный ответ на повреждение, включающий инфильтрацию нейтрофилами, продуцирующими токсичные свободные радикалы, усугубляющими повреждение ткани. Через 18—24 часа при световой микроскопии видны такие признаки коагуляционного некроза, как кариопикноз и слабо выраженная эозинофилия цитоплазмы. Эти ранние изменения представлены на рис. 7.5 и в таблице 7.2.
МАКРОСКОПИЧЕСКИЕ ИЗМЕНЕНИЯ
На макропрепарате не удается увидеть никаких изменений в течение 18—24 часов после окклюзии артерии, хотя некоторые методики окраски (в частности, тетразолием) позволяют идентифицировать зоны инфаркта и ранее. Наиболее часто ишемия и инфаркт начинаются с субэндокардиальных слоев и затем распространяются в стороны и по направлению к эпикарду.
Поздние изменения
Поздние патоморфологические изменения, характерные для дальнейшего течения острого инфаркта (табл. 7.2), включают: 1) очищение миокарда от некротических масс макрофагами и 2) отложение коллагена и формирование рубцовой ткани.
Таблица 7.2. Динамика патологических изменений при трансмуральном инфаркте
Время |
Событие |
Ранние изменения 1~2 мин 10 мин |
Падение уровня АТФ, нарушение сократимости Снижение уровня АТФ на 50%, отек клетки, снижение мембранного потенциала и повышение аритмической готовности |
20—24 мин 1-3 часа 4-12 часов |
Необратимое повреждение клетки Волнистость миофибрилл Кровоизлияния, отек, инфильтрация полиморфноядерными нейтрофилами |
Время |
Событие |
18-24 часа |
Коагуляционный некроз (пикнотические ядра с эозинофильной цитоплазмой), отек |
2—4 дня |
Тотальный коагуляционный некроз (отсутствуют ядра и исчерченность, вокруг — гиперемированная ткань); появление моноцитов |
Поздние изменения 5-7 дней |
Желтое размягчение вследствие резорбции отмерших тканей макрофагами |
|
7 дней + 7 недель |
Ремоделирование левого желудочка Фиброз и завершение рубцевания |
Необратимо поврежденные кардиомиоциты не регенерируют, они резорбируются и замещаются рубцовой тканью. Макрофаги проникают в воспаленный миокард вскоре после инфильтрации его нейтрофилами и удаляют некротическую ткань. Этот период резорбции ткани, когда наряду с мертвыми кардиомиоцитами разрушаются и удаляются элементы соединительной ткани, называется желтым размягчением. Активный фагоцитоз в сочетании с истончением и растяжением зоны инфаркта на этой стадии приводит к структурной слабости стенки желудочка и повышению риска ее разрыва. Впоследствии развивается фиброз и через 7 недель после инфаркта формирование рубца заканчивается (рис. 7.5).
Функциональные изменения
НАРУШЕНИЕ СОКРАТИМОСТИ
Инфаркт миокарда быстро приводит к снижению сократительной функции желудочка и уменьшению сердечного выброса. Далее сердечный выброс еще больше снижается из-за потери синхронности сокращения миоцитов: если инфаркт приводит к снижению сократимости, зона инфаркта характеризуется как «гипокинетичная», вообще не сокращающиеся сегменты называются «акинетичными», а «дискинетичные» сегменты в систолу выбухают наружу.
«ОГЛУШЕННЫЙ» И «СПЯЩИЙ» МИОКАРД
Ранее считалось, что ишемическое повреждение миокарда приводит либо к необратимому некрозу миокарда, либо к быстрому полному восстановлению функции кардиомиоцитов. Теперь стало известно, что ишемические атаки иногда могут приводить к длительной сократительной дисфункции без некроза кардиомиоцитов, и впоследствии их функция может полностью восстанавливаться.
Когда после периода тяжелой ишемии (но не некроза), даже после восстановления нормального коронарного кровотока, длительно сохраняется систолическая дисфункция, говорят об «оглушенном» (станнирующем) миокарде. В этом случае функциональные, биохимические и ультраструк-турные ишемические изменения обратимы, и сократимость постепенно восстанавливается. Механизм этого отсроченного восстановления функции пока не известен, но возможно он связан с перегрузкой кардиомиоцитов кальцием, накоплением свободных радикалов или электромеханической диссоциацией вследствие транзиторной дисфункции саркоплазматического ретикулума. В целом выраженность «оглушенности» пропорциональна степени предшествующей ишемии. Таким образом, «оглушенный» миокард, видимо, является патофизиологическим ответом на ишемическое повреждение, недостаточное для развития необратимого некроза.
Гибернирующий («спящий») миокард отличается от «оглушенного». Этим термином обозначают хроническую сократительную дисфункцию миокарда в условиях стойкого снижения коронарного кровотока, обычно при многососудистом поражении. В этой ситуации отсутствуют необратимые повреждения, и сократимость может восстанавливаться сразу после восстановления адекватного кровотока. В основе этого феномена лежит снижение сократительной активности миокарда в условиях хронической гипоперфузии с соответствующим балансом между низкой доставкой кислорода к миокарду и низкой его активностью (миокард как бы находится в «спячке»).
Концепции «оглушенного» и «спящего» миокарда имеют свое клиническое приложение. Например, если у пациента с острым инфарктом миокарда или хронической ишемической болезнью сердца выявляются зоны потенциально обратимого нарушения локальной сократимости, это может повлиять на решение о целесообразности реваскуляризации миокарда, в частности путем операции аортокоронарного шунтирования. В настоящее время возможности выявления «оглушенного» и «спящего» миокарда ограничены, однако для этого применяются некоторые радионуклидные методики (например, ПЭТ, описанная в главе 3).
РЕМОДЕЛИРОВАНИЕ ЛЕВОГО ЖЕЛУДОЧКА
После ИМ возникают изменения геометрии как поврежденной, так и интактной стенки левого желудочка. Изменения размеров камеры и толщины стенки влияют на долгосрочное состояние функции левого желудочка и прогноз.
В раннем постинфарктном периоде может произойти расширение зоны инфаркта, когда пораженный сегмент увеличивается в размерах без дополнительного некроза кардиомиоцитов. Расширение зоны инфаркта отражает истончение и дилатацию некротической ткани, видимо, за счет «соскальзывания» миофибрилл, в результате чего объем кардиомиоцитов в зоне инфаркта уменьшается. Расширение инфаркта может иметь негативные последствия, поскольку при этом увеличивается размер желудочка, а значит: 1) растет напряжение стенки, 2) нарушается систолическая сократительная функция, 3) повышается вероятность развития аневризмы.
Помимо раннего расширения зоны инфаркта, ремоделирование желудочка может включать дилатацию перегруженных неповрежденных сегментов, которые подвергаются повышенному напряжению стенки. Эта дилатация начинается в раннем постинфарктном периоде и затем продолжается в течение недель и месяцев. В начале дилатация полостей играет компенсаторную роль (она увеличивает сердечный выброс по закону Франка Старлинга), но прогрессирующее расширение желудочка может в конце концов привести к сердечной недостаточности и желудочковым аритмиям.
Неблагоприятное ремоделирование левого желудочка может быть предотвращено определенными вмешательствами. Например, в остром периоде инфаркта реперфузионная терапия позволяет ограничить размер инфаркта и, таким образом, уменьшить вероятность его расширения. Установлено также, что применение ингибиторов ангиотензин-превращающего фермента препятствует ремоделированию и снижает кратко- и долгосрочную смертность у постинфарктных больных (см. ниже).
ИНФАРКТЫ С Q- И БЕЗ Q-ЗУБЦА
Патоморфологически, как мы уже видели, инфаркты миокарда делятся на трансмуральные и субэндокардиальные. Ранее считалось правилом, что при трансмуральных инфарктах на ЭКГ после исходной элевации сегмента ST образуется патологический зубец Q, в то время как для субэндокардиальных характерна депрессия сегмента ST без последующего появления зубца Q. Теперь, однако, известно, что изменения ЭКГ не столь четко коррелируют с патанатомической картиной, и между этими двумя типами инфарктов имеется значительное количество промежуточных форм. В частности, у некоторых пациентов с трансмуральным инфарктом на ЭКГ отсутствуют Q-зубцы, в то время как при субэндокардиальных инфарктах, напротив, зубцы Q иногда присутствуют. В целом, однако, инфаркты без Q-зубца обычно отличаются от нфарктов с Q-зубцом меньшими размерами и вызываются неокклюзи-рующими тромбами (или более короткими периодами тяжелой ишемии).
В клиническом аспекте, тем не менее, целесообразно различать инфаркты с Q-зубцом и без Q-зубца. Ранняя внутрибольничная смертность у больных инфарктом без зубца Q ниже, однако на протяжении нескольких месяцев наблюдения у этих пациентов отмечается высокая частота повторных инфарктов и смертности, поскольку у них часто имеется тяжелое многососудистое поражение коронарного русла, и окружающая некроз жизнеспособная ткань подвергается повторным ишемическим эпизодам. Поэтому в этих группах больных для определения тактики лечения особенно важно достаточно активное обследование.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Субъективные и объективные симптомы острого инфаркта миокарда (таблица 7.3) непосредственно связаны с описанными ранее патофизиологическими процессами.
Таблица 7.3. Симптомы инфаркта миокарда
1. Характерный болевой синдром
2. Активация симпатической нервной системы |
|
3. Парасимпатические (вагусные) эффекты |
|
4. Воспалительный ответ |
• Невысокая лихорадка |
5. Изменения со стороны сердца |
|
6. Прочие |
|
Сопровождающая инфаркт миокарда боль по характеру напоминает стенокардию (см. главу 6), но обычно превышает ее по интенсивности, длится дольше и имеет более широкую иррадиацию. Подобно стенокардии, боль появляется вследствие воздействия на нервные окончания высвобождающихся из ишемизированных кардиомиоцитов медиаторов, таких как аденозин и лактат. Поскольку при инфаркте миокарда ишемия продолжается и приводит к некрозу, эти провоцирующие вещества продолжают накапливаться и воздействуют на афферентные нервные окончания более длительно. При инфаркте боль часто распространяется на другие области в пределах дерматомов С7-Т114, включая шею, плечи и руки. Боль при инфаркте миокарда развивается быстро и нередко практически сразу достигает максимума, и тогда у человека появляется сильный страх смерти. В отличие от приступа стенокардии, боль не ослабевает в покое, эффект нитроглицерина также нередко отсутствует.
Однако не все инфаркты миокарда сопровождаются болями. До 25% инфарктов миокарда протекают бессимптомно и диагностируются лишь ретроспективно при рутинном ЭКГ-обследовании или развитии осложнений. Это особенно часто происходит у диабетиков, которые вследствие периферической нейропатии могут не испытывать боли. Кроме того, пациенты с инфарктом миокарда, осложненным перикардитом, могут испытывать более острые, плевритоподобные (см. главу 14), а не типичные ангинозные боли.
Комбинация боли и недостаточной стимуляции барорецепторов (при наличии гипотензии) может вызвать выраженную реакцию симпатической нервной системы. Системными проявлениями выброса катехоламинов являются потливость, тахикардия и холодные липкие кожные покровы вследствие вазоконстрикции.
Из-за падения сократимости и, соответственно, ударного объема левого желудочка повышаются диастолические объем и давление. Повышение внутрижелудочкового давления, усугубляемое повышенной его жесткостью вследствие ишемии, передается на левое предсердие и легочные вены. Развивающийся в результате застой в легких снижает их эластичность и стимулирует юкстакапиллярные рецепторы. За счет этих J-рецепторов реализуется рефлекс, заключающийся в частом поверхностном дыхании, вызывающем субъективное чувство одышки. Транссудация жидкости внутрь альвеол еще более усиливает этот симптом.
При физикальном исследовании выявляются некоторые аускультативные признаки, зависящие от локализации и распространенности инфаркта. В течение короткого времени после инфаркта может выслушиваться IV тон, характеризующий сокращение предсердия в условиях диастолической дисфункции левого желудочка. У многих больных после инфаркта может выслушиваться III тон (фаза быстрого заполнения), что отражает нарушение систолической функции левого желудочка. При распространении воспаления на перикард может отмечаться шум трения перикарда. Наконец, при дисфункции или инфаркте папиллярных мышц, вызывающих недостаточность митрального клапана, или при разрывах межжелудочковой перегородки могут появляться систолические шумы.
Некроз миокарда также активирует системный ответ на воспаление. В ответ на повреждение тканей макрофаги и сосудистый эндотелий продуцируют цитокины — интерлейкин-1 и фактор некроза опухоли. Эти медиаторы ответственны за ряд клинических проявлений, таких как умеренная лихорадка и лейкоцитоз.
ДИАГНОСТИКА
Диагноз острого инфаркта миокарда основывается на: 1) клинической картине и данных анамнеза, 2) типичных изменениях ЭКГ и 3) определении кардиоспецифичных макромолекул в сыворотке крови. В таблице 7.4 перечислены другие причины острой боли в грудной клетке, похожие на боли при инфаркте миокарда.
Таблица 7.4. Дифференциальный диагноз острого ИМ
Состояния |
Характерные черты |
|
Патология сердца Инфаркт миокарда |
|
Перикардит |
|
Диссекция аорты |
|
|
Патология легких Тромбоэмболия легочной артерии |
|
Пневмония |
|
Пневмоторакс |
|
|
Патология ЖК тракта Спазм пищевода |
|
Изменения ЭКГ
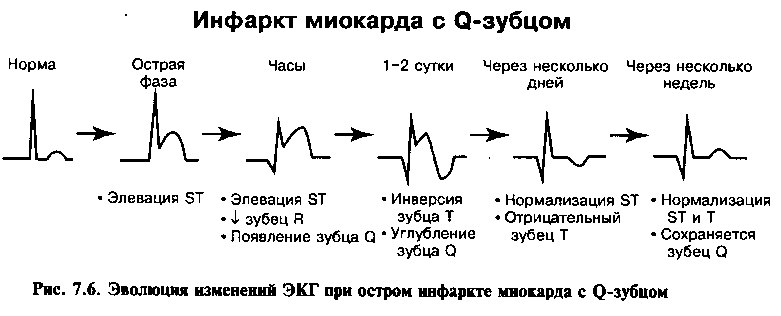
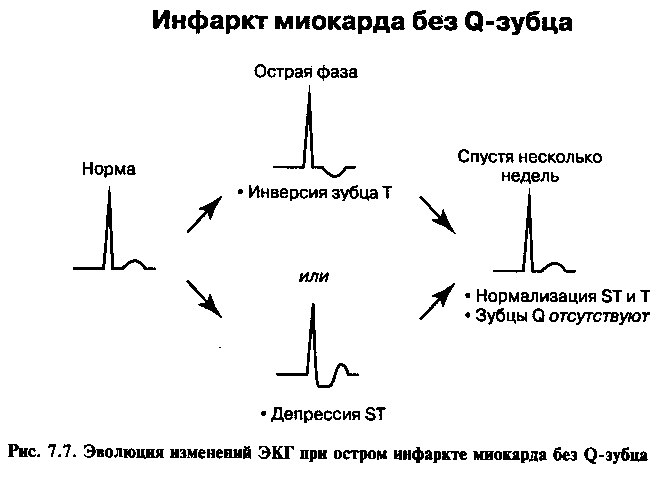
Изменения на ЭКГ развиваются в типичной для инфаркта временной последовательности, описанной в главе 4. При инфарктах с зубцом Q в отведениях, соответствующих зоне некроза, отмечаются элевация сегмента ST, инверсия зубца Т и зубцы Q (рис. 7.6), в то время как при инфарктах без Q-зубца имеются депрессия сегмента ST и инверсия зубца Т (рис. 7.7).
Состояния |
Характерные черты |
Острый холецистит |
|
Сывороточные маркеры инфаркта
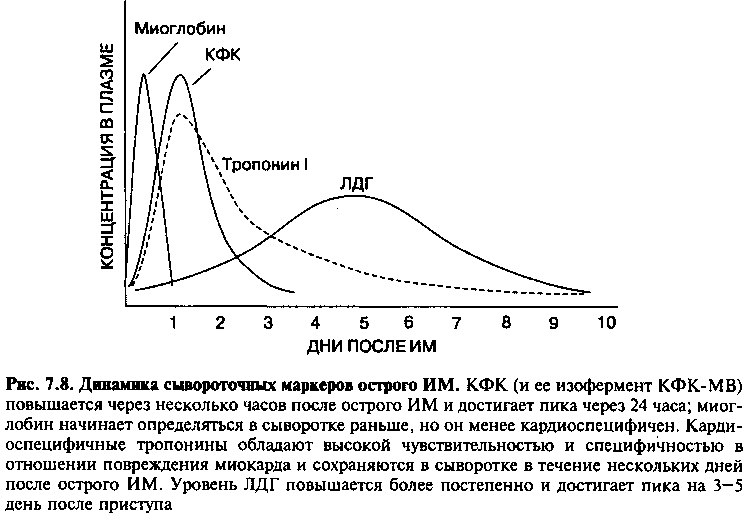
Некроз миокардиальной ткани ведет к разрывам сарколеммы, и внутриклеточные макромолекулы попадают сначала в интерстициальное пространство, а затем в кровоток (рис. 7.8). Лабораторные методы выявления этих молекул в сыворотке крови играют важную роль в диагностике инфаркта миокарда.
Креатинфосфокиназа. Фермент креатинфосфокиназа (КФК) обратимо переносит фосфатную группу с креатинфосфата, эндогенной резервной формы макроэргических фосфатов, на АДФ, образуя таким образом АТФ. Поскольку креатинфосфокиназа содержится в сердце, скелетных мышцах, мозге и многих других органах, ее концентрация в сыворотке может повышаться после повреждения любой из этих тканей.
Однако КФК имеет три изофермента, что улучшает диагностическую специфичность в отношении миокарда: КФК-MM (содержится в основном в скелетных мышцах), КФК-ВВ (доминирует в мозговой ткани) и КФК-MB (характерна для миокарда). Оценка динамики КФК-MB в сыворотке сейчас является «золотым стандартом» ферментной диагностики ИМ; современные моноклональные тест-системы для этого изофермента обладают высокой чувствительностью и специфичностью. Необходимо отметить, что сердце также содержит КФК-MM, так что во время острого инфаркта миокарда уровень этого изофермента также повышается. Более того, небольшие количества КФК-MB обнаруживаются в других тканях, включая матку, простату, кишечник, диафрагму и язык. В отсутствие
травм этих органов повышение КФК-МВ высоко специфично для повреждения миокарда. Поскольку КФК-МВ составляет 1%—3% от всей КФК скелетной мускулатуры, мышечная травма или внутримышечные инъекции также могут вызывать появление в крови этого изофермента. Поэтому для уточнения диагноза инфаркта миокарда принято вычислять отношение: КФК-МВ/общая КФК. При использовании чувствительного моноклонального тестового набора для КФК-МВ это отношение обычно составляет > 2,5% в случае повреждения миокарда и менее 2,5%, если источником является только скелетная мускулатура.
Уровень КФК-МВ в сыворотке начинает повышаться через 4—8 часов после инфаркта, достигает пика через 24 часа и возвращается к норме через 48—72 часа (рис. 7.8). Эта временная последовательность очень важна, так как КФК-МВ из других источников (например, поврежденых скелетных мышц) или при другой патологии сердца (например, при миокардите) обычно не подчиняется этому закону. Реперфузия (например, в результате тромболитической терапии) при инфаркте миокарда приводит к быстрому вымыванию фермента и более раннему пику КФК и КФК-МВ.
Поскольку уровень КФК-МВ в первые несколько часов от начала инфаркта повышается незначительно, нормальное его значение при однократном определении в приемном отделении больницы не исключает инфаркта миокарда. Поэтому уровень КФК-МВ не может использоваться для принятия решения, кого из пациентов с болью в грудной клетке следует госпитализировать для дальнейшего наблюдения, а кого можно отпустить домой. В настоящее время подобное решение принимается исходя из анамнеза, физикальных данных и ЭКГ.
Для лучшей диагностики инфаркта миокарда в критические первые часы после начала приступа было предложено определять несколько других сывороточных маркеров. Например, были описаны изоформы КФК-МВ. КФК-МВ2 высвобождается из зоны инфаркта миокарда и, поступая в кровь, подвергается ферментативному превращению в КФК-МВ]. По последним данным, в первые часы после развития симптоматики абсолютное повышение КФК-МВ2 (или повышенное значение отношения МВ2 /МВ]) в два раза чувствительнее в диагностике острого ИМ, чем стандартное определение КФК-МВ. Миоглобин, гемсодержащий белок, попадает в кровоток при повреждении миокарда и может определяться в сыворотке через 2 часа после начала ИМ, то есть намного раньше, чем повышается уровень КФК-МВ. Однако быстрое удаление этой молекулы почками и ее низкая специфичность для повреждения миокарда ограничивают ее диагностическую ценность. Наиболее перспективными являются недавно разработанные тест-системы для определения кардиоспецифичных изоформ тропонинов Т и I. Каждый из этих маркеров обладает высокой специфичностью в отношении инфаркта миокарда, и они могут выявляться в крови через 3 часа после начала болевого синдрома. В результате их высокой чувствительности и специфичности использование тропонинов в диагностике острого ИМ быстро растет.
Лактатдегидрогеназа (ЛДГ) катализирует обратимую реакцию образования лактата из пирувата. ЛДГ содержится во многих тканях и имеет пять изоформ. Наиболее специфичным для сердца изоферментом является ЛДГ] и величина отношения ЛДГ! / ЛДГ2 >1,0 указывает на некроз миокарда. (ЛДГ2 содержится в эритроцитах, ЛДГ4 и ЛДГ5 обнаруживаются в печени и скелетных мышцах.) Поскольку ЛДГ достигает своего пика на 3—5 сутки после ИМ, определение этого фермента диагностически значимо для пациентов, поступающих в стационар в эти сроки, когда повышение КФК уже прошло.
Если симптоматика и результаты стандартного лабораторного обследования неоднозначны, для диагностики острого ИМ могут быть полезны некоторые другие методы. Эхокардиография может выявить появившиеся нарушения локальной сократимости в зоне инфаркта. Она может также помочь в диагностике механических осложнений инфаркта, таких как дефект межжелудочковой перегородки или митральная регургитация. Сцинтиграфия миокарда с технецием-99т пирофосфатом может подтвердить наличие некротизированной ткани: пирофосфат накапливается в зонах с высокой концентрацией кальция, что в частности характерно для инфаркта миокарда. Через 12 часов после инфаркта миокарда очаг инфаркта выявляется как зона повышенной интенсивности радиоактивного излучения (см. главу 3).
ЛЕЧЕНИЕ В ОСТРОМ ПЕРИОДЕ
В раннем периоде инфаркта лечение направлено на поддержание жизнеспособности ишемизированного миокарда. Современное лечение инфаркта претерпело революционные изменения при появлении методик реперфузии, способность которых ограничивать некроз и улучшать выживаемость хорошо доказана.
Тромболитическая терапия
Поскольку большинство острых Q-инфарктов миокарда развиваются вследствие окклюзирующего внутрикоронарного тромбоза, тромболитические агенты могут восстановить кровоток и ограничить повреждение миокарда. Используемые сейчас тромболитические препараты включают стрептокиназу, анизоилированный плазминоген-стрептокиназный активированный комплекс (АПСАК) и рекомбинантный тканевой активатор плазминогена (ТАП). Хотя механизмы их действия различны (рис. 7.9), все эти средства активируют протеолитический фермент плазмин, который растворяет фибриновый тромб. Введение тромболитика в первые несколько часов от начала острого ИМ в большинстве случаев восстанавливает коронарный кровоток и существенно ограничивает объем повреждения. Наиболее успешна ранняя тромболитическая терапия. В частности, при введении тромболитика в течение первого часа Q-ИМ реперфузия достигается в 80% случаев, что сопровождается значительным снижением смертности.
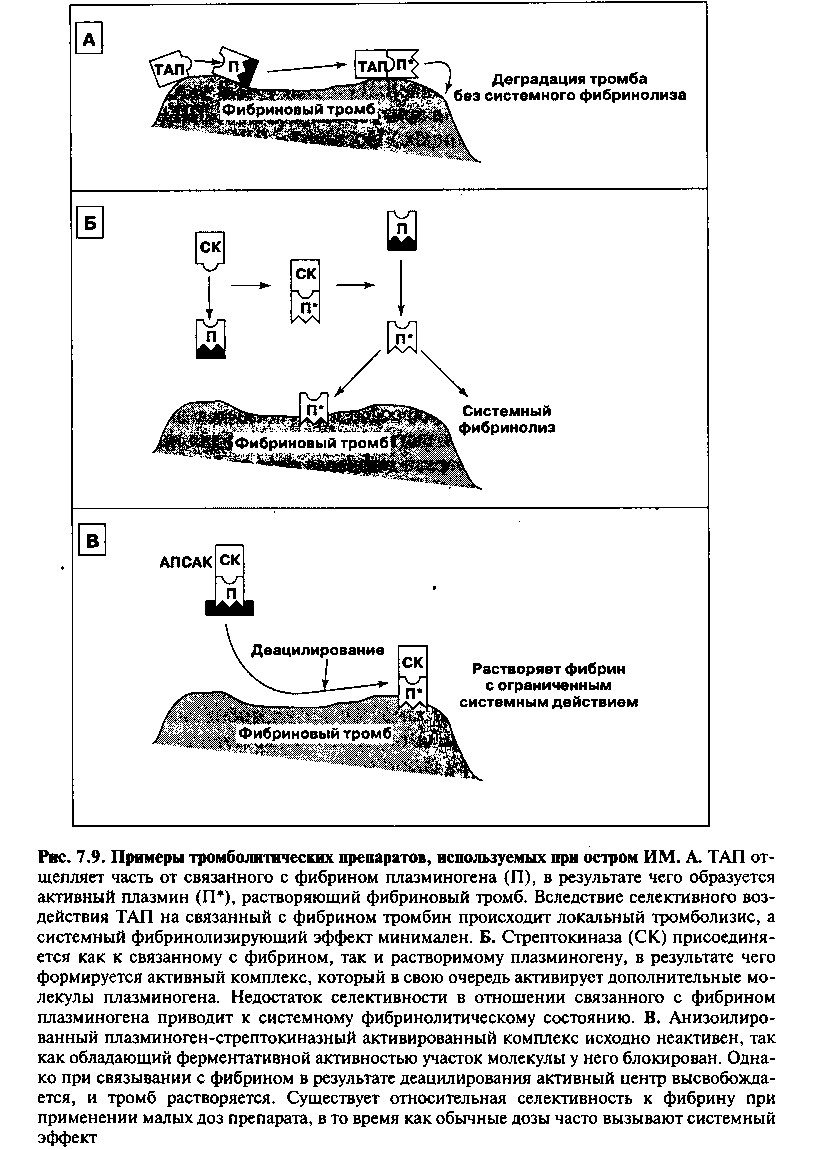
Проведены крупномасштабные исследования, оценивавшие сравнительную эффективность тромболитических препаратов. Хотя в первых работах не было обнаружено разницы в выживаемости после применения имевшихся препаратов, проведенное недавно крупное исследование (GUSTO) показало, что ТАЛ обладает некоторым преимуществом перед стрептокиназой, хотя он несколько чаще вызывает развитие инсультов. Однако по современным представлениям, большее значение имеет не выбор тромболити-ка, а сроки, в которые он вводится: при начале тромболизиса в первые 2 часа от начала болей смертность в два раза меньше, чем после 6 часов.
Противопоказаниями к тромболизису являются состояния, при которых имеется риск кровотечений после произведенного тромболизиса (например, при обострении язвенной болезни, сопутствующем геморрагическом диатезе, в раннем послеоперационном периоде).
При успешном восстановлении коронарного кровотока в результате тромболизиса сегмент ST возвращается к изолинии и пик концентрации сывороточной КФК наступает раньше, чем обычно (так как ткань не подвергается некрозу, и фермент «вымывается» в результате восстановления кровотока). После тромболизиса для предотвращения немедленной реокклюзии сосуда обычно применяется внутривенная антикоагулянтная терапия гепарином.
Первичная ЧТКА
Альтернативой тромболитической терапии при остром ИМ является неотложная катетеризация сердца с проведением чрескожной транслюминальной коронарной ангиопластики («первичной ЧТКА») инфарктс-вязанной артерии. ЧТКА имеет особенно важное значение у больных с противопоказаниями к тромболизису. Имеющиеся данные свидетельствуют, что немедленная ангиопластика может восстановить коронарный кровоток у более чем 90% пациентов и снизить частоту повторной ишемии, рецидивирующих инфарктов и смертность. Однако в настоящее время первичная ЧТКА выполняется только в центрах с очень большим опытом ангиопластики и оборудованных для проведения ее в экстренном порядке.
Стационарное лечение
Независимо от проведения тромболизиса или первичной ЧТКА, лечение ИМ в остром периоде направлено на 1) восстановление баланса между доставкой кислорода к миокарду и потребностью в нем с целью предотвращения повторной ишемии, 2) купирование боли и 3) профилактику и лечение возможных осложнений. Все это наилучшим образом достигается при госпитализации больного в отделение коронарной патологии (ОКП) — специализированное отделение больницы, предназначенное для лечения острого ИМ и его осложнений. Стандартное лечение включает:
1) Постельный режим в спокойной обстановке, при наличии постоянного мониторирования ЭКГ для своевременного выявления нарушений ритма.
2) Оксигенотерапия через маску или носовые катетеры с целью максимальной доставки кислорода.
3) Аспирин снижает агрегацию тромбоцитов и, как хорошо доказано, уменьшает смертность и частоту рецидивов инфарктов. Лечение аспирином следует начинать немедленно после начала острого ИМ и при отсутствии противопоказаний (например, геморрагический диатез или аллергия) продолжать пожизненно.
4) (3-блокаторы. В отсутствие противопоказаний (астма, бронхоспазм, гипотензия) p-блокаторы должны назначаться всем больным, так как было показано, что они снижают частоту рецидивов инфаркта и повторной ишемии. Уменьшая симпатические влияния на сердце, р-блока-торы уменьшают нагрузку на миокард и способствуют его электрической стабильности.
5) Нитраты купируют ангинозный приступ посредством дилатации вен, а следовательно — снижения потребйости миокарда в кислороде из-за уменьшения венозного возврата к сердцу (4- преднагрузка, и поэтому 4-напряжение стенки). Хотя нитраты ранее считались основным классом препаратов для лечения хронической ишемической болезни сердца, их влияние на уровень смертности не установлено, и сейчас их применение в терапии острого ИМ ограничивается симптоматической помощью при стенокардии (и в качестве вазодилататора у некоторых больных сердечной недостаточностью или высокой гипертензией на ранних сроках инфаркта). Поэтому при остром ИМ нитроглицерин часто назначают сублингвально или, иногда, внутривенно до полного купирования болей, а затем препарат отменяют.
6) Морфин — мощный анальгетик, применяемый для купирования болей и уменьшения тревожности и, таким образом, снижающий тонус симпатической нервной системы и потребность миокарда в кислороде. Морфин также является венодилататором и таким образом снижает преднагрузку, что в свою очередь способствует снижению потребности миокарда в кислороде.
7) Антикоагулянты. Кроме случаев применения антикоагулянтов в качестве адьювантной терапии после тромболизиса, внутривенное введение гепарина показано также пациентам с большими передними инфарктами, низким сердечным выбросом или мерцанием предсердий с целью профилактики тромбоза полостей сердца. Всем госпитализированным по поводу острого ИМ пациентам следует также вводить подкожно небольшие дозы гепарина для профилактики тромбоза глубоких вен нижних конечностей и малого таза.
8) Ингибиторы ангиотензинпревращающего фермента (АПФ) (описаны далее в главе 17) продемонстрировали свою способность предотвращать постинфарктное ремоделирование желудочка, снижать частоту развития сердечной недостаточности и постинфарктной стенокардии, улучшать выживаемость. Они дополняют положительное действие аспирина и p-блокаторов. Показано, что ингибиторы АПФ вызывают наиболее выраженное улучшение прогноза у пациентов с постинфарктной систолической дисфункцией левого желудочков (то есть со сниженной фракцией выброса левого желудочка). Поэтому пероральный прием ингибиторов АПФ начинают на ранних сроках ИМ, и при наличии снижения сократительной функции ЛЖ продолжают неопределенно долго после выписки из стационара.
ОСЛОЖНЕНИЯ
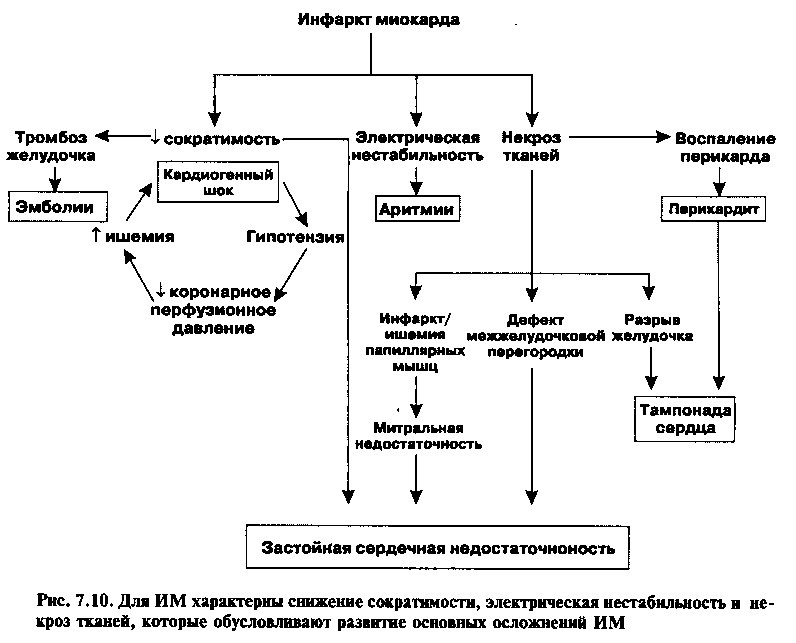
В основе осложнений острого инфаркта миокарда лежат воспаление, механические и электрические нарушения, возникающие в зонах некроза миокарда (рис. 7.10). Ранние осложнения развиваются из-за наличия самого некроза. Те же осложнения, которые развиваются спустя несколько дней или недель, связаны с воспалением и процессами замещения некротической ткани.
..........i Эмболии
Кардиогенный шок
Т ишемия
Постинфарктная стенокардия и рецидивирующий инфаркт миокарда
Постинфаркгная стенокардия отмечается у 20%—30% пациентов и отражает неадекватность остаточного коронарного кровотока. Она является плохим прогностическим признаком и коррелирует с повышенным риском рецидива инфаркта. Таких пациентов следует немедленно отправлять на коронароангиографию, за которой нередко следует реваскуляризация, то есть ЧТКА или АКШ.
У 5%—20% больных в течение первых 6 недель инфаркт рецидивирует. Рецидив инфаркта может развиться в той же зоне, что и первичный инфаркт (при условии наличия жизнеспособной ткани) или в ранее интактном сегменте.
Аритмии
Нарушения ритма сердца в острую фазу инфаркта миокарда встречаются очень часто (таблица 7.5) и являются основной причиной смерти. Современные отделения коронарной патологии (ОКП) во многом ориентированы именно на выявление и лечение нарушений ритма, так что аритмическая смертность у госпитализированных больных относительно невысока. Патогенетические механизмы аритмий при ИМ следующие:
1. Прекращение перфузии элементов проводящей системы (то есть СА узла, АВ узла, ножек пучка Гиса); источники кровоснабжения проводящей системы сердца в норме перечислены в таблице 7.6.
2. Локальное накопление метаболитов (например, клеточный ацидоз) и изменения трансмембранной концентрации ионов из-за нарушения проницаемости мембран.
3. Стимуляция вегетативной нервной системы (симпатической и парасимпатической).
4. Назначение потенциально аритмогенных препаратов (например, дигиталиса, допамина).
Таблица 7.5. Аритмии при остром инфаркте миокарда
Ритм |
Причина |
Синусовая брадикардия |
|
Синусовая тахикардия |
|
Ритм |
Причина |
НЖЭС, фибрилляция пред- |
• зсн |
сердий |
• Ишемия предсердий |
ЖЭС, ЖТ, ФЖ |
|
АВ блокада I, II, III степени |
|
ЖЭС — желудочковая экстрасистола, ЖТ — желудочковая тахикардия, ФЖ — фибрилляция желудочков, НЖЭС — наджелудочковые экстрасистолы
ФИБРИЛЛЯЦИЯ ЖЕЛУДОЧКОВ
Большая часть внезапных сердечных смертей во время острого ИМ происходит вследствие фибрилляции желудочков (быстрой, беспорядочной электрической активности желудочков). К сожалению, большая часть летальных исходов наступает до приезда скорой помощи и госпитализации. Эпизоды фибрилляции желудочков в первые 48 часов часто связаны с транзиторной электрической нестабильностью и не влияют на долгосрочный прогноз у переживших ее больных. Однако при развитии ФЖ в более поздние сроки острого ИМ фибрилляция обычно отражает тяжелую дисфункцию левого желудочка и ассоциируется с последующей высокой смертностью.
Желудочковая экстрасистолия, желудочковая тахикардия и фибрилляция желудочков во время острого ИМ имеют в своей основе либо механизм ре-ентри, либо повышение автоматизма кардиомиоцитов желудочков (см. главу И). Желудочковая экстрасистолия встречается часто и обычно не нуждается в лечении, если не является групповой, политопной или частой. Лидокаин (антиаритмик I класса, см. главу 17) при внутривенном введении эффективно предупреждает развитие фибрилляции желудочков в условиях ишемии, но не применяется для рутинного лечения всех больных ИМ из-за потенциальных побочных эффектов, а также в связи с готовностью персонала ОК.П оказать помощь при появлении нарушений ритма.
НАДЖЕЛУДОЧКОВЫЕ НАРУШЕНИЯ РИТМА
При остром ИМ также часто встречаются наджелудочковые аритмии. Синусовая брадикардия возникает из-за стимуляции блуждающего нерва или ишемии СА узла, обычно при нижней локализации инфаркта. Синусовая тахикардия также встречается часто и может иметь несколько причин, включая боль и тревожность, застойную сердечную недостаточность, действие лекарств (например, допамина, нитратов) или уменьшение объема циркулирующей крови (гиповолемию). Дифференциальная диагноста-ка между сердечной недостаточностью и гиповолемией может потребовать трансвенозной катетеризации легочной артерии: снижение давления заклинивания легочной артерии будет указывать на гиповолемию, а повышение — на недостаточность кровообращения. Поскольку синусовая тахикардия увеличивает потребность сердца в кислороде и может провоцировать ишемию миокарда, очень важно установить и ликвидировать ее причину. Наджелудочковая экстрасистолия и фибрилляция предсердий (см. главу 12) могут развиваться из-за ишемии предсердий или их растяжения при левожелудочковой недостаточности.
НАРУШЕНИЯ ПРОВОДИМОСТИ
Нарушения проводимости (АВ блокада и блокады ножек пучка Гиса) часто развиваются в остром периоде ИМ. Причиной их могут быть ишемия и некроз элементов проводящей системы (таблица 7.6) или, в случае АВ блокад, транзиторное повышение тонуса блуждающего нерва (обсуждается в гл. 11). Вагусная активность увеличивается вследствие стимуляции афферентных нервных окончаний в зоне воспаления или генерализованной активации вегетативной нервной системы при болевом синдроме.
Таблица 7.6. Источники кровоснабжения проводящей системы сердца
Элементы проводящей системы |
Первичный источник кровоснабжения |
|
СА узел АВ узел Пучок Гиса Правая ножка пучка Гиса Левая ножка пучка Гиса: Левая передняя ветвь Правая задняя ветвь |
|
СА — синоатриальный; АВ — атриовентрикулярный, ПКА — правая коронарная артерия, ПМЖА — передняя межжелудочковая артерия, ЗМЖА — задняя межжелудочковая артерия.
Дисфункция миокарда
ЗАСТОЙНАЯ СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Острая ишемия приводит к нарушению сократимости желудочка (снижению систолической функции), а также к повышению жесткости его стенки (нарушение диастолической функции); оба этих обстоятельства могут приводить к появлению симптомов сердечной недостаточности. Кроме того, ЗСН могут провоцировать аритмии и остро развившиеся механические осложнения ИМ (рис. 7.10). Симптомы декомпенсации включают одышку, хрипы в легких и III тон при аускультации. Лечение соответствует стандартам терапии ЗСН (рассматриваются в главе 9) и включает диуретики и вазодилататоры.
КАРДИОГЕННЫЙ ШОК
Кардиогенный шок развивается при инфаркте миокарда, охватывающем более 40% массы миокарда левого желудочка, и характеризуется резким снижением сердечного выброса и артериальной гипотензией (систолическое АД < 90 мм рт. ст.) и, как следствие, неадекватной перфузией периферических тканей. Он также может сопутствовать описанным ниже тяжелым механическим осложнениям. При кардиогенном шоке возникает своеобразный порочный круг (рис. 7.10): 1) гипотензия приводит к снижению коронарной перфузии и усилению ишемического повреждения и 2) снижение ударного объема увеличивает размеры левого желудочка и, следовательно, потребность миокарда в кислороде. Несмотря на агрессивное лечение, летальность при кардиогенном шоке превышает 70%.
Пациентам при кардиогенном шоке показано внутривенное введение инотропных агентов (например, добутамина) с целью повышения сердечного выброса и артериальных вазодилататоров для снижения постнагрузки на левый желудочек. Часто стабилизации состояния удается достичь путем внутриаортальной баллонной контрпульсации. Применяемое для этого устройство вводится в аорту через бедренную артерию и состоит из эластичного баллона, который расширяется в диастолу, повышая давление в аорте и улучшая перфузию коронарного русла и периферических тканей. Спадение баллона в систолу присасывает кровь из левого желудочка в аорту. Из-за риска тромбоза и суперинфекции этот метод может использоваться только в течение нескольких дней.
Инфаркт правого желудочка
Приблизительно у одной трети больных с инфарктом нижней стенки левого желудочка также развивается некроз части правого желудочка, поскольку у большинства людей эти зоны имеют единый источние кровоснабжения (обычно ПКА). Развивающееся вследствие этого снижение сократимости и эластичности правого желудочка приводит к появлению симптомов декомпенсации по большому кругу (например, к набуханию яремных вен), не соответствующих по выраженности явлениям левожелудочковой недостаточности. Поскольку в результате дисфункции правого желудочка нарушается приток крови к легким и снижается наполнение левого желудочка, может развиться тяжелая артериальная гипотензия. В этих условиях стабилизировать АД можно путем внутривенной инфузии больших объемов жидкости при инвазивном контроле гемодинамики путем катетеризации легочной артерии.
Механические осложнения
Механические осложнения ИМ обусловлены ишемией и некрозом тканей.
РАЗРЫВ ПАПИЛЛЯРНОЙ МЫШЦЫ
Ишемический некроз и разрыв папиллярной мышцы левого желудочка может быстро привести к летальному исходу из-за остро развившейся тяжелой митральной регургитации. Частичный разрыв, при котором регургитация менее выражена, не всегда заканчивается летально, однако может привести к развитию сердечной недостаточности или отека легких. Задне-медияльная папиллярная мышца левого желудочка более подвержена некрозу, чем передне-латеральная, в связи с более частыми нарушениями ее кровоснабжения, это осложнение более типично для нижне-заднего ИМ.
РАЗРЫВ СВОБОДНОЙ СТЕНКИ ЖЕЛУДОЧКА
Разрыв свободной стенки ЛЖ в области некроза является нечастым, но смертельным осложением, которое может развиться в первые две недели после ИМ. Он чаще встречается у женщин и пациентов с артериальной гипертензией в анамнезе. Развивающееся в результате такого разрыва кровотечение в полость перикарда быстро ведет к тампонаде сердца, то есть к нарушению заполнения сердца из-за скопления крови в перикарде (см. главу 14). Выживают при этом единицы.
Иногда при неполном разрыве свободной стенки, прикрытом тромбом, формируется псевдоаневризма сердца. Эту ситуацию можно сравнить с бомбой замедленного действия, поскольку в любой момент может произойти полный разрыв с гемотампонадой. При своевременном выявлении (обычно с помощью эхокардиографии) хирургическая коррекция может предотвратить неблагоприятный исход.
РАЗРЫВ МЕЖЖЕЛУДОЧКОВОЙ ПЕРЕГОРОДКИ
Это осложнение аналогично разрыву свободной стенки желудочка, за тем исключением, что патологический поток крови не попадает в перикард. Тем не менее, кровь поступает через дефект перегородки из левого в правый желудочек, что обычно вызывает развитие сердечной недостаточности из-за объемной перегрузки малого круга кровообращения. При аускультации часто выслушивается громкий систолический шум вдоль левого края грудины, отражающий сброс крови через перегородку. Этот шум можно дифференцировать от остро возникшей митральной регургитации с помощью доплер-эхокардиографии или путем измерения сатурации кислорода в крови, взятой из правых камер сердца с помощью центрального венозного катетера. При сбросе оксигенированной крови из левого желудочка в правый содержание кислорода в правом желудочке становится выше, чем в правом предсердии.
ИСТИННАЯ АНЕВРИЗМА ЛЕВОГО ЖЕЛУДОЧКА
Истинная аневризма является поздним осложнением, возникающим через несколько недель или месяцев после ИМ. Она развивается в тех случаях, когда стенка желудочка под действием фагоцитоза некротических масс ослабляется, но не разрывается, так что при сокращении неповрежденного миокарда этот участок локально выбухает наружу. В отличие от описанной выше псевдоаневризмы, в этом случае сообщение между полостью левого желудочка и перикардом отсутствует, так что разрыва и тампонады сердца не происходит. Осложения аневризмы левого желудочка включают: 1) внутрижелудочковый тромбоз из-за замедленного кровотока, являющийся потенциальным источником периферических тромбоэмболий, 2) желудочковые аритмии из-за растяжения кардиомиоцитов и 3) сердечную недостаточность из-за снижения сердечного выброса, поскольку часть ударного объема ЛЖ во время систолы «теряется» в полости аневризмы.
Признаками наличия аневризмы левого желудочка являются сохраняющаяся элевация сегмента ST в соответствующих отведениях ЭКГ (спустя недели после острого ИМ с Q-зубцом) и выбухание стенки ЛЖ при рентгенографии органов грудной клетки. Наличие аневризмы (и ее тромбоза) обычно может быть подтверждено с помощью эхокардиографии.
Перикардит
Острый перикардит может развиться в раннем постинфарктном периоде, когда некроз и нейтрофильная инфильтрация распространяются на эпи- и перикард (см. главу 14). При этом могут отмечаться острая боль, лихорадка и шум трения перикарда, который помогает отличить перикардит от ишемической боли. Перикардит является относительным противопоказанием для назначения антикоагулянтов, поскольку существует риск кровотечения из воспаленного перикарда. Частота развития перикардита при ИМ снизилась после появления тромболитической терапии, так как этот вид лечения ограничивает зону некроза и воспаления.
Тромбоэмболии
Турбулентность и замедленный кровоток в зонах нарушенной сократимости могут провоцировать внутриполостное тромбообразование, особенно при верхушечной локализации инфаркта или при формировании истинной аневризмы. Последующие тромбоэмболии могут приводить к развитию инфарктов других органов (например, эмболии церебральных сосудов приводят к инсультам).
СТРАТИФИКАЦИЯ РИСКА И ТАКТИКА
ведения больных, перенесших им
Большинство больных могут быть выписаны уже через 5—7 дней после ИМ. Тем не менее, возможны рецидивы инфаркта, и очень важно своевременно выявлять пациентов с высоким риском этого осложнения. Наиболее важным прогностическим фактором после ИМ является выраженность дисфункции левого желудочка. Другими факторами, указывающими на неблагоприятный прогноз, являются ранняя постинфарктная стенокардия, большой объем жизнеспособного миокарда в зоне нарушенного кровотока и желудочковые аритмии высоких градаций.
С целью идентификации больных с высоким риском применяется проба с физической нагрузкой на тредмиле (перед выпиской применяются нагрузки низкой интенсивности, а через 4—6 недель — субмаксимальный тест). Пациенты, у которых при нагрузке выявляются значительные нарушения, а также больные с ранней постинфарктной стенокардией подлежат коронароангиографии для оценки характера поражения коронарного русла.
Стандартная терапия после выписки включает 1) аспирин, 2) р-блока-тор, 3) ингибитор АПФ (особенно при наличии дисфункции левого желудочка). Гиполипидемическая терапия (наиболее часто ингибитором ГМГ-КоА-редукгазы, то есть статином) показана для достижения целевого уровня ЛНП < 100 мг/дл. Необходимо обратить внимание на другие факторы риска сердечно-сосудистых заболеваний (курение, артериальная гипертензия, сахарный диабет), а также на стандартную программу физической реабилитации, которая обычно ускоряет выздоровление.
ЗАКЛЮЧЕНИЕ:
1. Инфаркт миокарда (ИМ) представляет собой некроз миокарда вследствие длительной ишемии. Большая часть инфарктов развивается вследствие окклюзирующего тромбоза коронарной артерии в месте атеросклеротической бляшки. Обычно триггерным фактором для развития тромбоза является разрыв бляшки, запускающий активацию тромбоцитов и каскад гемокоагуляции. Атеросклеротическая дисфункция эндотелия усугубляет этот процесс из-за сниженной секреции вазодилатирующих и антитромботических веществ.
2. Острый ИМ приводит к ряду биохимических и механических нарушений, которые снижают сократимость левого желудочка, его эластичность (нарушение диастолической функции) и предрасполагают к развитию опасных для жизни нарушений ритма. Инфаркт запускает механизм воспалительного ответа, который приводит к замещению некротических масс рубцовой тканью. Транзиторная тяжелая ишемия без формирования инфаркта может приводить к появлению «оглушенного» миокарда, феномена, при котором систолическая дисфункция сохраняется и после купирования ишемии, а затем сократимость постепенно восстанавливается.
3. Диагностика острого ИМ основана на типичной клинической картине, характерных изменениях ЭКГ и преходящем повышении кардиоспе-цифических ферментов в крови.
4. В остром периоде ИМ основными принципами лечения являются: ограничение объема повреждения миокарда путем реперфузии с помощью тромболитических препаратов или экстренной чрескожной ангиопластики. Также обычно назначаются аспирин, 0-блокаторы, ингибиторы АПФ и, при необходимости, гепарин. Для купирования болей применяются морфин и нитраты. В ОКП проводится мониторирование ЭКГ с целью своевременного выявления аритмий.
5. Непосредственными осложнениями ИМ являются такие нарушения ритма, как желудочковая тахикардия и фибрилляция желудочков, АВ блокады, блокады ножек пучка Гиса и суправентрикулярные аритмии. Вследствие дисфункции левого желудочка или механических осложнений (разрыв свободной стенки левого желудочка или межжелудочковой перегородки, остро возникшая митральная регургитация) могут развиваться кардиогенный шок или застойная сердечная недостаточность. Кроме того, нарушения сократимости в зоне инфаркта могут предрасполагать к образованию тромбов.
6. Стандартная терапия после выписки из стационара включает аспирин и 0-блокатор, а также, при необходимости, ингибитор АПФ (при наличии дисфункции левого желудочка), непрямые антикоагулянты (при наличии внутрижелудочковых тромбов или большой зоны акинеза) и гиполипидемические препараты.
7. Стратификация риска после ИМ направлена на выявление пациентов с высоким риском рецидива инфаркта и смерти. Систолическая дисфункция левого желудочка, электрическая нестабильность (желудочковые нарушения ритма высоких градаций) и признаки ишемии во время нагрузочной пробы являются неблагоприятными прогностическими факторами и требуют соответствующего дополнительного обследования и коррекции терапии.