Атеросклероз
Атеросклероз — это заболевание артерий мышечного типа, при котором их внутренний слой утолщается за счет липидных отложений и фиброзной ткани. Это состояние, часто обозначаемое как «уплотнение артерий», нередко поражает коронарные и церебральные артерии, приводя к инфарктам миокарда и инсультам. В результате атеросклероз является основной причиной большинства смертей в развитых странах, в частности — в США он обусловливает более половины всей смертности за год. Несмотря на высокую распространенность и длительное изучение (пато-морфологические основы известны уже более ста лет), механизмы развития атеросклероза остаются до конца не раскрытыми.
Эта глава описывает патоморфологию атеросклеротических поражений, факторы риска атеросклероза; особое внимание уделяется клеточным и биохимическим процессам, которые могут играть роль в его развитии.
АРТЕРИАЛЬНАЯ СТЕНКА
В норме стенка артерий мышечного типа состоит из трех слоев (рис. 5.1): интимы (она ближе всего к просвету сосуда и поэтому «интимно» связана с кровью), медии (среднего слоя) и наружной адвентиции.
Интима представлена одним слоем эндотелиальных клеток, под которыми находится соединительная ткань. Эндотелий формирует барьер, который удерживает циркулирующую кровь внутри просвета сосуда и в норме выполняет множество дополнительных метаболических и сигнальных функций, которые помогают поддерживать целостность сосудистой
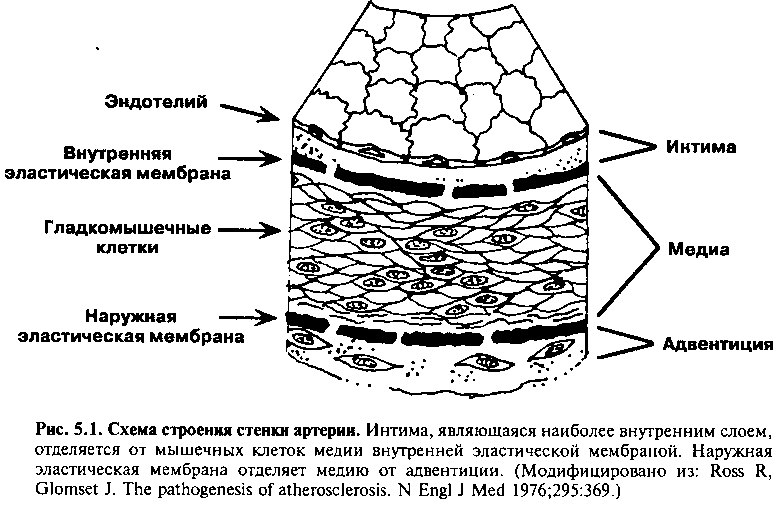
стенки. В случае развития атеросклеротических поражений они формируются внутри интимы.
Медия — наиболее толстый слой нормальной артериальной стенки, который отделяется от интимы и адвентиции соответственно внутренней и наружной эластическими мембранами, имеющими поры, через которые могут проникать клетки. Медия состоит в основном из гладкомышечных клеток и матрикса из коллагена, эластина и протеогликанов. В крупных артериях эластического типа, таких как аорта и ее крупные ветви, медия в систолу растягивается под влиянием высокого давления, создаваемого сердцем, а затем в диастолу сокращается, так что кровь проталкивается вперед на протяжении всего кардиоцикла. В меньших по размеру артериях мышечного типа степень сокращения гладкомышечных клеток внутри медии определяет сопротивление сосуда и, таким образом, регулирует кровоток.
Наружный слой стенки артерии, адвентиция, содержит фибробласты и коллаген, а также кровеносные сосуды (vasa vasorum), нервы и лимфатические сосуды артерии. Считается, что адвентиция не участвует в развитии атеросклероза.
АТЕРОСКЛЕРОТИЧЕСКИЕ ПОРАЖЕНИЯ
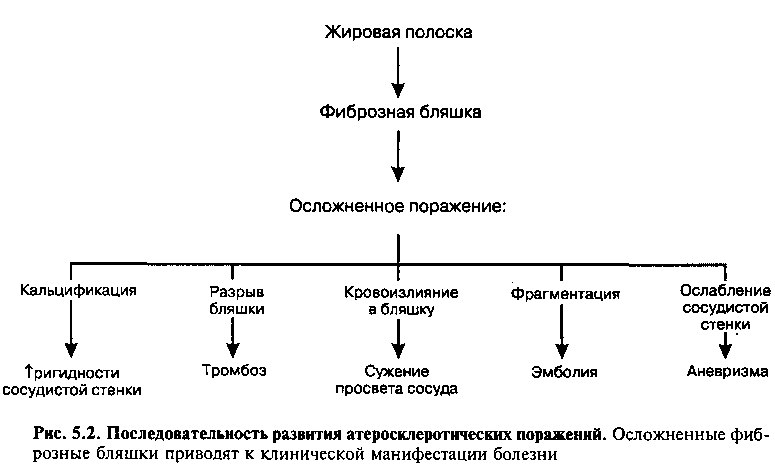
Патоморфологическим субстратом атеросклероза являются жировые полоски и фиброзные бляшки. Рис. 5.2 суммирует последовательность событий, в результате которых эти поражения ведут к клинической манифестации заболевания.
Жировые полоски
Жировые полоски являются наиболее ранними атеросклеротическими поражениями, которые можно увидеть. На макроскопическом уровне они представлены участками желтого прокрашивания внутренней поверхности артерии. Они часто выглядят как пятна менее 1 мм в диаметре или как полоски, достигающие 1—2 мм в ширину и до 1 см в длину (рис. 5.3). Они не выступают в просвет артерии и не мешают кровотоку. Под микроскопом жировые полоски характеризуются субэндотелиальным накоплением крупных «пенистых клеток», заполненных внутриклеточными липидами (рис. 5.4). С использованием моноклональных антител было продемонст-

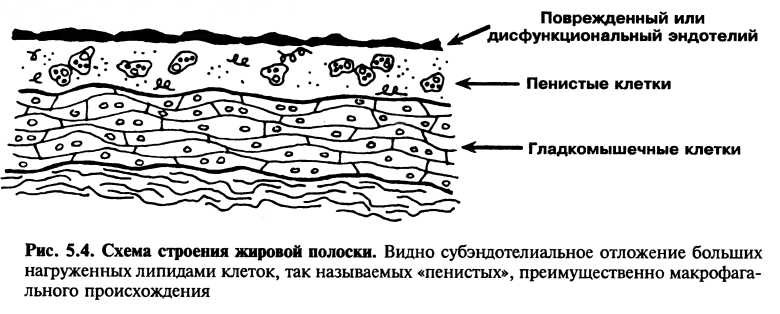
рировано, что большинство пенистых клеток в жировых полосках произошли от макрофагов, а небольшая их часть — от гладкомышечных клеток.
Жировые полоски начинают развиваться с раннего возраста и видны в аорте и коронарных артериях большинства лиц до двадцати лет. Они бессимптомны, а при некоторых локализациях могут со временем претерпевать обратное развитие. Однако при других локализациях, таких как коронарное русло, жировые полоски, видимо, являются предшественниками более опасных фиброзных бляшек.
Фиброзная бляшка
Фиброзные бляшки — это более далеко зашедшая стадия атеросклеротического процесса, обусловливающая развитие клинических проявлений болезни. Судя по всему, фиброзные бляшки развиваются из жировых полосок и локализуются в коронарных артериях и других сосудах в тех же самых местах, где наиболее типично встречаются жировые полоски. На макропрепарате фиброзная бляшка (рис. 5.5) представляет собой плотный светло-серый возвышающийся элемент. Она может выбухать в просвет сосуда и, при достаточных размерах, создавать клинически значимый стеноз, ограничивающий кровоток по сосуду.
Гистологически большая часть связанных с формированием фиброзной бляшки изменений артериальной стенки происходит в интиме, а именно аккумуляция моноцитов, лимфоцитов, пенистых клеток и соединительной ткани. В отличие от жировых полосок, в фиброзных бляшках большинство пенистых клеток гладкомышечного происхождения. Фиброзные бляшки часто содержат некротическое ядро, состоящее из клеточного детрита, дегенерирующих пенистых клеток и кристаллов холесте-
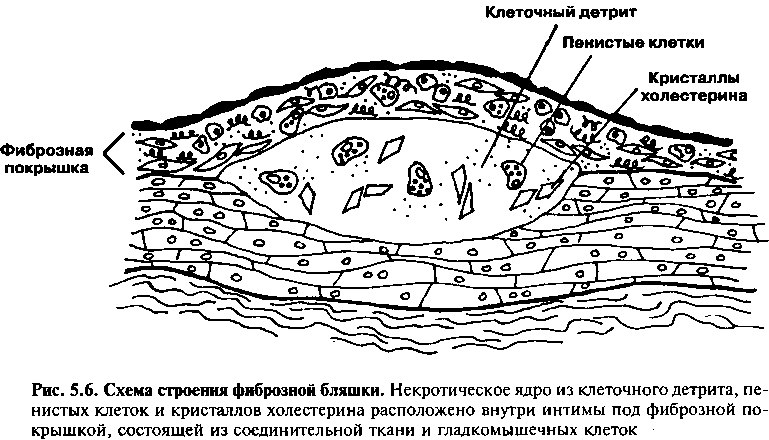
рина, они отделены от просвета артерии фиброзной покрышкой, в основном состоящей из экстрацеллюлярного соединительнотканного матрикса с включенными в него гладкомышечными клетками (рис. 5.6).
У больных атеросклерозом фиброзные бляшки распределены в сосудистом русле неравномерно. Они чаще всего встречаются в брюшной аорте, затем с убывающей частотой — в коронарных артериях, подколенных артериях, нисходящей части грудной аорты, внутренних сонных артериях и сосудах Виллизиева круга. Области, кровоснабжаемые этими артериями, наиболее часто страдают от последствий атеросклероза.
Клиническое значение фиброзных бляшек определяется последствиями, к которым приводит уменьшение кровотока по артерии или нарушение целостности сосудистой стенки (рис. 5.2). Эти осложнения могут развиваться в результате:
1. Кальцификации фиброзной бляшки, приводящей к ригидности сосудистой стенки и повышению ее ломкости.
2. Разрыва или изъязвления фиброзной бляшки, в результате чего тромбогенные субстанции контактируют с кровотоком, вызывая образование тромба в месте повреждения. В результате тромбоза может произойти окклюзия сосуда и, как следствие, инфаркт миокарда или инсульт. Иногда тромботические массы включаются в состав бляшки, увеличивая ее размер.
3. Кровоизлияния в фиброзную бляшку в результате разрыва фиброзной покрышки или мелких капилляров, кровоснабжающих бляшку. Образующаяся гематома может усугублять сужение просвета сосуда.
4. Эмболизации дистального отдела артерии фрагментами разорвавшейся атеромы.
5. Уменьшения прочности сосудистой стенки, поскольку фиброзная бляшка подвергает соседнюю медию повышенному давлению, что может спровоцировать атрофию и потерю эластической ткани, с последующей дилатацией артерии (то есть формированием аневризмы).
Клиническое значение этих осложнений описывается позже, в главах
6, 7 и 15.
РАСПРОСТРАНЕННОСТЬ И ФАКТОРЫ РИСКА
Выяснив, кого поражает атеросклероз, мы можем кое-что узнать о его патогенезе. Уже давно было известно, что распространенность клинически выраженного атеросклероза наиболее высока в обществах с большим потреблением мяса, и гораздо ниже в популяциях, придерживающихся диеты с низким содержанием жиров. Например, смертность от коронарной болезни сердца исторически в Японии всегда была гораздо ниже, чем в США. Этот факт, возможно, связан с диетическими различиями, поскольку исследования японцев, переехавших в США и сменивших диету и образ жизни, показали значительное повышение уровня общего холестерина и коронарной смертности.
Несколько крупных эпидемиологических исследований в США и в Европе выявили ряд особенностей и привычек, коррелирующих с развитием атеросклероза и ишемической болезни сердца. Одним из таких исследований является Фрамингемское, в котором жители городка в штате Массачусетс на протяжении нескольких десятилетий наблюдались на предмет выявления взаимосвязи между определенными факторами риска и развитием коронарной болезни. Другое исследование — Multiple Risk Factor Intervention Trial (MRFIT), в котором более 325000 мужчин обследовались для выявления связи между факторами риска и последующей сердечно-сосудистой заболеваемостью и смертностью. С помощью этих исследований удалось установить четыре основных, потенциально модифицируемых, фактора риска, ассоциированных с атеросклерозом: 1) аномальные уровни липидов в крови («дислипидемия»), 2) артериальная гипертензия, 3) курение, 4) сахарный диабет.
Также было выявлено несколько основных немодифицируемых факторов риска, в том числе пожилой возраст, мужской пол, развитие ИБС у родственников в молодом возрасте (то есть, моложе 55 лет у родственников мужского пола и моложе 65 лет у родственников женского пола). Дополнительные потенциально модифицируемые факторы риска включают ожирение, сидячий образ жизни и эмоциональные стрессы. Повышенный уровень аминокислоты гомоцистеина в крови также недавно был признан независимым фактором риска, о чем будет сказано ниже.
Дислипидемия
Целый ряд факторов свидетельствует, что анормальный уровень липидов в крови является основным фактором риска развития атеросклероза.
Исследования показали, что в обществах с относительно высоким уровнем общего холестерина характерен наивысший уровень смертности от ишемической болезни сердца. Например, в странах с низким потреблением насыщенных жиров и низким уровнем сывороточного холестерина (к ним относятся Япония и некоторые страны Средиземноморья) смертность от ИБС ниже, чем в США и других странах с более высоким потреблением жиров и уровнем холестерина. Данные Фремингемского и многих других исследований также свидетельствуют о том, что риск ишемической болезни сердца растет с повышением уровня холестерина. У лиц с общим холестерином 240 мг/дл риск ИБС приблизительно в два раза выше, чем при уровне холестерина 200 мг/дл. Однако при исследовании различных форм липидов крови и их функций оказалось, что не всякий холестерин является «плохим».
ПУТИ ТРАНСПОРТА ХОЛЕСТЕРИНА
В кровотоке переносчиками липидов являются липопротеины. Они состоят из липидного ядра, окруженного растворимыми фосфолипидами и свободным холестерином, а также апопротеинами, которые отвечают за направление липопротеинов к специфическим органам и тканевым рецепторам. Известно пять основных классов липопротеинов, различающихся по плотности, липидному составу и аполипопротеинам (табл. 5.1).
Таблица 5.1. Липопротеины плазмы (в порядке возрастания плотности)
Тип |
Источник |
Основные липидные компоненты |
Ассоциированные * апопротеины |
Хиломикроны |
ЖК тракт |
Триглицериды |
A-I, А-П, A-IV, В-48, С-1, С-П, С-Ш, Е |
ЛОНП |
Печень |
Триглицериды |
В-100, С-1, С-П, С-Ш, Е |
ЛПП |
Остатки ЛОНП |
Холестерин |
В-100, Е |
ЛНП |
Метаболизм ЛПП |
Холестерин |
В-100 |
ЛВП |
Печень, ЖК тракт |
Холестерин |
A-I, А-П, С-1, С-П, С-Ш, Е |
Рис. 5.7 дает характеристику основных путей метаболизма циркулирующих липопротеинов. Жиры, поступившие с пищей, включаются в цикл, известный как экзогенный путь. Пищевые холестерин и триглицериды всасываются в кишечнике, включаются в хиломикроны клетками кишечного эпителия и транспортируются через лимфатические протоки в венозную систему. Эти большие, богатые триглицеридами частицы гидролизируются ферментом липопротеинлипазой, которая высвобождает жирные кислоты, захватываемые периферическими тканями, такими как жировая и мышечная. Образующиеся остатки хиломикронов состоят преимущественно из холестерина. Эти остатки поглощаются печенью, которая затем выделяет липиды в виде свободного холестерина либо желчных кислот обратно в кишечник.
Эндогенный путь начинается с того, что липопротеины очень низкой плотности (ЛОНП) высвобождаются из печени в кровоток. Хотя основным липидным компонентом ЛОНП являются триглицериды, содержа-
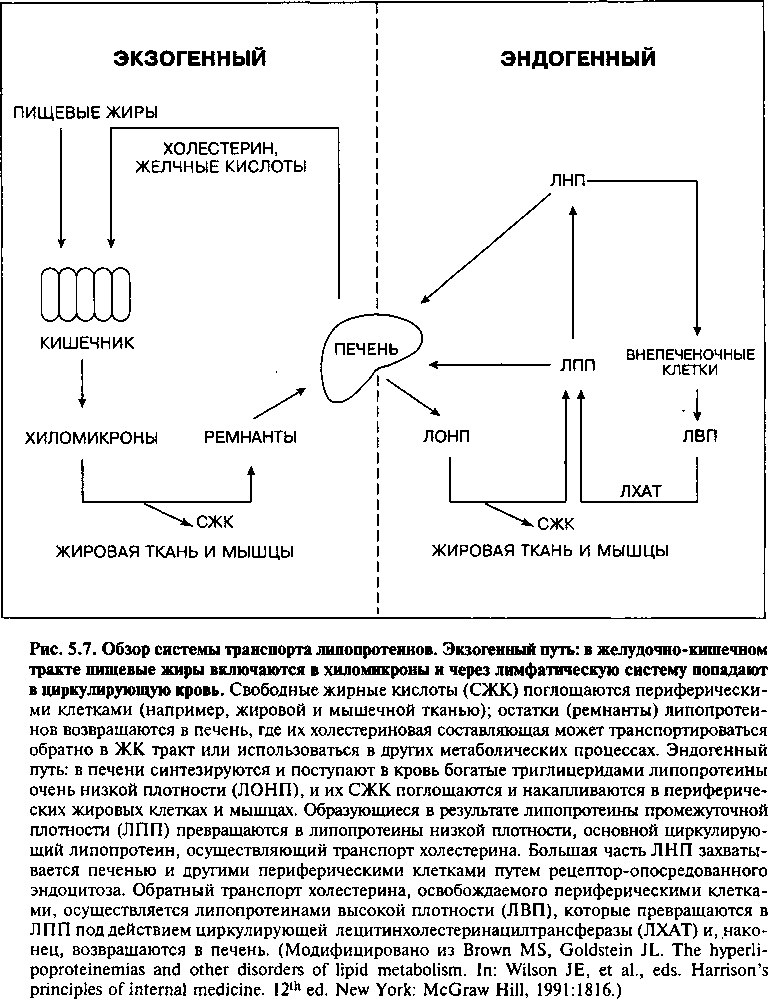
щие мало холестерина, основная часть холестерина поступает из печени в кровь именно в составе ЛОНП. Липопротеинлипаза мышечных клеток и жировой ткани отщепляет от ЛОНП свободные жирные кислоты, которые проникают в клетки, а циркулирующий остаток липопротеина, называемый ремнантным липопротеином промежуточной плотности (ЛПП), содержит в основном эфиры холестерина. Дальнейшие преобразования, которым ЛПП подвергается в крови, ведут к появлению богатых холестерином частиц липопротеинов низкой плотности (ЛНП). Приблизительно 75% циркулирующих ЛНП захватываются печенью и внепеченочными клетками благодаря наличию ЛНП-рецепторов. Остаток подвергается деградации отличными от классического ЛНП-рецепторного пути способами, в основном посредством моноцитарных клеток-мусорщиков.
Считается, что холестерин, поступающий в кровь из периферических тканей, транспортируется липопротеинами высокой плотности (ЛВП) в печень, где он вновь включается в липопротеины или секретируется в желчь (путь, включающий ЛПП и ЛНП, называется обратным транспортом холестерина). Таким образом, ЛВП, видимо, играет защитную роль в отношении отложения липидов в атеросклеротических бляшках. В крупных эпидемиологических исследованиях уровень циркулирующего ЛВП обратно коррелирует с развитием атеросклероза. Поэтому ЛВП часто называют «хорошим» холестерином в противоположность «плохому» холестерину ЛНП.
Семьдесят процентов холестерина плазмы транспортируется в виде ЛНП, и повышенный уровень ЛНП тесно коррелирует с развитием атеросклероза. В конце 1970-х гг. докторами Брауном и Гольдштейном была продемонстрирована центральная роль рецептора ЛНП в доставке холестерина к тканям и его клиренсе из кровотока. Экспрессия рецепторов ЛНП регулируется механизмом отрицательной обратной связи: нормальный или высокий уровень внутриклеточного холестерина подавляет экспрессию рецептора ЛНП на уровне транскрипции, в то время как снижение внутриклеточного холестерина повышает экспрессию рецептора с последующим увеличением захвата ЛНП клеткой. Пациенты с генетическими дефектами рецептора ЛНП (обычно гетерозиготы с одним нормальным и одним дефектным геном, кодирующим рецептор) не могут эффективно удалять ЛНП из кровотока, что приводит к высокому уровню ЛНП в плазме и склонности к преждевременному развитию атеросклероза. Это состояние называется семейной гиперхолестеринемией. Гомозиготы с полным отсутствием рецепторов ЛНП встречаются редко, но у этих людей инфаркт миокарда может развиваться уже в первое десятилетие жизни.
Недавно на основании различий в плотности и плавучести были идентифицированы подклассы ЛНП. Лица с более мелкими и плотными частицами ЛНП (свойство, определяемое как генетическими, так и внешними факторами) подвержены более высокому риску инфаркта миокарда, чем обладатели менее плотных разновидностей. Пока остается неясным, почему более плотные частицы ЛНП сопряжены с большим риском, однако это может быть связано с большей подверженностью плотных частиц окислению, ключевому моменту атерогенеза, о чем будет сказано ниже.
Возрастает число свидетельств того, что триглицериды сыворотки, в основном транспортирующиеся в составе ЛОНП и ЛПП, могут также играть важную роль в развитии атеросклеротических поражений. Пока не ясно, является ли это их прямым действием или объясняется тем, что уровень триглицеридов обычно находится в обратном соотношении с уровнем ЛВП. Сахарный диабет, начинающийся во взрослом возрасте, является одним из частых клинических состояний, ассоциированных с гипертриглицеридемией и низким уровнем ЛВП, а часто — и с ожирением и артериальной гипертензией. Этот набор факторов риска, который может быть связан с инсулинорезистентностью (обсуждается в главе 13), особенно атерогенен.
ПРЕИМУЩЕСТВА ЛЕЧЕНИЯ ДИСЛИПИДЕМИИ
Многочисленные недавние исследования показали, что у больных ИБС снижение сывороточного холестерина с помощью диеты или лекарств может предотвратить прогрессирование атеросклеротических поражений коронарного русла. Имеются данные даже об умеренном регрессе стенозов коронарных артерий у пациентов, принимавших определенные гиполипидемические препараты. И, что более важно, было показано, что снижение холестерина ЛНП под действием лечения приводит к достоверному снижению частоты инфарктов миокарда и смертности, не пропорциональной пользе, ожидаемой от уменьшения степени стеноза артерии. Наибольшая польза была продемонстрирована от применения лекарств, которые конкурентно ингибируют гидроксиметил гл утарил-КоА-редукта-зу (ГМГ-КоА-редуктазу), фермент, определяющий скорость синтеза холестерина. Считается, что эти препараты, значительно снижающие уровень циркулирующего ЛНП, улучшают прогноз за счет уменьшения липидного содержимого атеросклеротических бляшек, уменьшая их склонность к разрыву. Отчетливая клиническая польза от снижения холестерина согласуется с предполагаемой патогенетической ролью холестерина ЛНП в развитии атеросклероза.
Современные национальные рекомендации предполагают скрининговое определение холестерина у всех взрослых. Для общей популяции желательным является уровень общего холестерина менее 200 мг/дл, холестерина ЛНП <130 мг/дл и холестерина ЛВП > 35 мг/дл. Лицам, не достигающим этих целевых уровней, рекомендуется диета и часто медикаментозное лечение, особенно при наличии основных факторов риска развития атеросклероза. Пациентам с диагностированной коронарной болезнью или после-перенесенного инфаркта рекомендуются даже более низкие целевые уровни ХС ЛНП (< 100 мг/дл).
ПОВЫШЕННЫЙ УРОВЕНЬ ЛИПОПРОТЕИНА (А)
КАК ФАКТОРА РИСКА АТЕРОСКЛЕРОЗА
Был выявлен еще один независимый фактор риска, связанный с липопротеинами, а именно, липопротеин (а). Липопротеин (а), уровень которого в первую очередь определяется генетически, подобен частице ЛНП, однако связан дисульфидным мостиком с особым апопротейном — так называемым апо(а). Апо(а) имеет структурное сходство с плазминогеном, плазменным белком, отвечающим за эндогенный лизис фибриновых тромбов (см. главу 7). Считается, что негативный эффект повышения липопротеина (а) связан с ингибированием нормальной тромболитической активности плазминогена.
ЭФФЕКТЫ ЭСТРОГЕНОВ
До менопаузы у женщин ИБС развиваются реже, чем у мужчин. Однако в постменопаузе коронарный риск у обоих полов уравнивается, и сердечно-сосудистые заболевания являются ведущей причиной смертности у женщин, значительно опережая другие причины, включая рак груди. Нормальный уровень эстрогенов в сыворотке снижает ЛНП и липопротеин (а) и повышает уровень ЛВП, что может отчасти объяснить защитную роль эстрогенов в отношении поражения коронарного русла. Эстрогены обладают также потенциально полезными антиоксидантным и антитром-боцитарным действием.
Поскольку уровень эстрогенов после менопаузы снижается, женщинам в постменопаузе часто назначается гормональная заместительная терапия, особенно при наличии гиперхолестеринемии и других факторов риска. Однако заместительная терапия эстрогенами может увеличивать риск развития рака эндометрия. Добавление прогестинов уменьшает риск развития рака этой локализации и, по-видимому, не уменьшает положительных эффектов чистых эстрогенов.
Курение
Курение — это фактор риска сердечно-сосудистых заболеваний, который, возможно, легче всего устранить. Многочисленные исследования показали увеличение риска атеросклероза, ишемической болезни сердца и внезапной смерти у курильщиков. Относительный риск смерти от ИБС в разных работах оценивается между 1,35 и 2,40 у всех курильщиков и между 1,43 и 3,50 у злостных курильщиков. Курение является независимым фактором риска и может также усугублять сердечно-сосудистые ос-ложения, ассоциированные с другими известными факторами риска.
Количество никотина и других химических веществ, попадающих в организм курильщика, очень вариабельно, так что трудно четко связать количество выкуриваемых сигарет с риском атеросклероза. Однако ясно, что даже минимальное по интенсивности курение повышает риск, и что наибольшей опасности подвергаются наиболее злостные курильщики. Специальные работы показали также, что курение сигарет с низким содержанием смол и никотина не снижает существенно риск инфаркта миокарда по сравнению с обычными сигаретами.
Точно не известно, почему курение сигарет приводит к атеросклеротическим осложнениям и коронарной болезни сердца. Потенциальные механизмы включают: снижение уровня ЛВП, неадекватную стимуляцию симпатической нервной системы никотином, замещение связанного с гемоглобином кислорода на оксид углерода, повышение агрегации тромбоцитов и дисфункцию эндотелия, вызываемую компонентами табачного дыма.
К счастью, некоторые из этих путей обратимы. Эпидемиологические исследования показали, что у людей, бросивших курить, заболеваемость коронарной болезнью сердца снижается по сравнению с продолжающими. В одной из работ было показано, что после трех лет отказа от курения риск развития коронарной болезни уравнивается с риском для людей, которые никогда не курили.
Гипертензия
Повышенное артериальное давление (как систолическое, так и диастолическое) является фактором риска развития атеросклероза, ИБС и инсульта (см. главу 13). Не существует специфического порога, после которого повышенное давление приводит к сердечно-сосудистой патологии; скорее существует ряд степеней риска, связанных с прогрессивно повышающимся давлением. Механизм, благодаря которому артериальная гипертензия вносит свой вклад в атерогенез, не ясен. Однако в экспериментах на животных было показано, что повышенное артериальное давление повреждает эндотелий и может увеличивать проницаемость сосудистой стенки для липопротеинов.
Снижение артериального давления с помощью диеты или лекарств снижает риск инсульта и, в меньшей степени, коронарных осложнений. Распространено мнение, что именно диагностика и лечение артериальной гипертензии способствовали 57% снижению смертности от инсультов и 50% снижению коронарной смертности, произошедшим в США в последние 20 лет.
Сахарный диабет
Диабет также является фактором риска атеросклероза, однако измерить этот риск сложно, поскольку при диабете широко распространены и другие факторы риска, такие как артериальная гипертензия и дислипидемия. Считают, что повышение риска связано у диабетиков с гликозилированием липопротеинов (что может увеличивать захват холестерина макрофагами-мусорщиками, как будет рассказано ниже) или с типичной для этого заболевания повышенной агрегацией тромбоцитов. До сих пор не установлено, снижает ли у диабетиков строгий контроль гликемии риск атеросклеротических поражений, как это имеет место при микросо-судистых осложнениях этого заболевания (например, ретинопатии).
Гомоцистеинемия
В добавление к перечисленным выше традиционным факторам риска недавние исследования показали достоверную связь между уровнем аминокислоты гомоцистеина в крови и заболеваемостью коронарным, церебральным и периферическим атеросклерозом. Риск инфаркта миокарда у лиц с наиболее высокими уровнями гомоцистеина по сравнению с пациентами с наиболее низкими уровнями повышается приблизительно в три раза. Механизм, с помощью которого гомоцистеин повышает риск атеросклероза, не известен, однако по имеющимся данным аномально высокий уровень этой аминокислоты может повреждать эндотелиальные клетки артериальной стенки. Обогащение рациона фолиевой кислотой и другими витаминами группы В может снижать уровень гомоцистеина, однако неизвестно, снижает ли подобное лечение коронарный риск.
КЛЕТОЧНЫЕ ЭЛЕМЕНТЫ СОСУДИСТОЙ СТЕНКИ
Для развития атеросклеротических поражений необходимо участие ключевых клеток артериальной стенки. Наиболее важны эндотелиальные клетки интимы артерии и сосудистые гладкомышечные клетки.
Эндотелиальные клетки
Клетки эндотелия выстилают интиму кровеносных сосудов и выполняют в норме весьма важные структурные, метаболические и паракринные функции (табл. 5.2). Во-первых, они формируют барьер, удерживающий циркулирующую кровь внутри просвета сосуда. Эндотелиальные клетки плотно прилегают друг к другу, ограничивая проникновение крупных молекул из кровотока в субэндотелиальное пространство.
Во-вторых, эндотелий препятствует тромбообразованию посредством выработки противотромботических поверхностных молекул (например, гепаран сульфата, тромбомодулина и активаторов плазминогена, см. главу 7) и высвобождения антитромбоцитарных факторов, включая простациклин и эндотелий-зависимый релаксирующий фактор. Последняя субстанция была идентифицирована как оксид азота или его очень близкое производное, и далее будет именоваться ЭЗФР-NO. Эффекты этих ключевых медиаторов описаны в главе 6.
В-третьих, клетки эндотелия секретируют вазоактивные субстанции, которые непосредственно влияют на сократимость гладкомышечных клеток в подлежащей медии. В нормальной артерии эффекты определенных сосудорасширяющих веществ (например, простациклина и ЭЗФР-NO) играют ведущую роль в расслаблении гладких мышц и расширении диаметра сосуда.
Наконец, путем синтеза определенных веществ (таких как гепаран сульфат и ЭЗФР-NO) эндотелиальные клетки ингибируют миграцию и пролиферацию гладкомышечных клеток, что, как мы увидим далее, также может играть важную антиатеросклеротическую роль. Таким образом, в норме эндотелиальный слой интимы представляет собой защитную ан-титромбогенную поверхность, метаболически активен и синтезирует вазоактивные вещества.
Таблица 5.2. Функции клеток эндотелия
Функция |
Нормальный эндотелий |
«Поврежденный» эндотелий |
Барьерная функция |
Формирует плотный барьер, препятствующий проникновению крупных молекул и клеток в субэндотелиальное пространство |
Демонстрирует повышенную проницаемость |
Антитромбо-тическая активность |
Противостоит тромбозу благодаря действию гепаран сульфата, тромбомодулина, активаторов плазминогена и секреции ингибиторов тромбоцитов (простациклина, ЭЗФР-NO) |
Снижение антитромботи-ческих свойств (например, уменьшение секреции простациклина и ЭЗФР-NO) |
Влияние на тонус сосудов |
Способствует вазодилатации благодаря секреции простациклина и ЭЗФР-NO |
Способствует вазоконстрикции из-за нарушения секреции простациклина и ЭЗФР-NO |
Влияние на гладкомышечные клетки |
Ингибирует миграцию и пролиферацию гладкомышечных клеток (через гепаран сульфат и ЭЗФР-NO) |
Способствует миграции и пролиферации гладкомышечных клеток (сниженная секреция ЭЗФР-NO, повышение секреции ТФР) |
ЭЗФР-NO — эндотелий зависимый фактор релаксации — оксид азота; ТФР — тромбоцитарный фактор роста
Напротив, при повреждении или дисфункции эндотелиальных клеток их активность изменяется в сторону, которая может способствовать атеросклерозу (табл. 5.2). Во-первых, проницаемость поврежденного эндотелиального слоя может увеличиваться, и в результате клетки не могут дольше служить барьером для миграции клеток и макромолекул из кровотока в субэндотелиальное пространство. Во вторых, поврежденные эндотелиальные клетки могут терять свои антитромботические свойства из-за сниженной продукции простациклина и ЭЗФР-NO. В-третьих, снижение секреции вазодилататоров (например, простациклина и ЭЗФР-NO) нарушает расслабление гладкомышечных клеток медии и приводит к относительной вазоконстрикции. В-четвертых, поврежденные эндотелиальные клетки секретируют повышенное количество митогенных соединений, таких как тромбоцитарный фактор роста (PDGF), который привлекает гладкомышечные клетки внутрь интимы и стимулирует их размножение. Наконец, поврежденный эндотелий секретирует факторы хемотаксиса, которые привлекают внутрь интимы другие ключевые клетки, включая циркулирующие моноциты, которые вовлекаются в развитие атеросклеротических поражений, о чем будет сказано ниже.
Гладкомышечные клетки сосудов
По современным данным ключевым событием в атерогенезе является пролиферация гладкомышечных клеток внутрь интимы артерии; эти мышечные клетки первично исходят из медии. Стимулы, приводящие к миграции и пролиферации этих клеток, будут описаны ниже.
Гладкомышечные клетки внутри сосудистой стенки проявляют фенотипическую гетерогенность как в норме, так и в патологии (рис. 5.8). В нормальной артерии гладкомышечные клетки «сократительного» фенотипа содержат большое количество сократительных белков, которые несут функцию укорочения миофибриллл и сокращения клетки. Гладкомышечные клетки в этом состоянии экспрессируют рецепторы ко многим вазоактивным субстанциям, таким как ангиотензин II, катехоламины, эндоте-
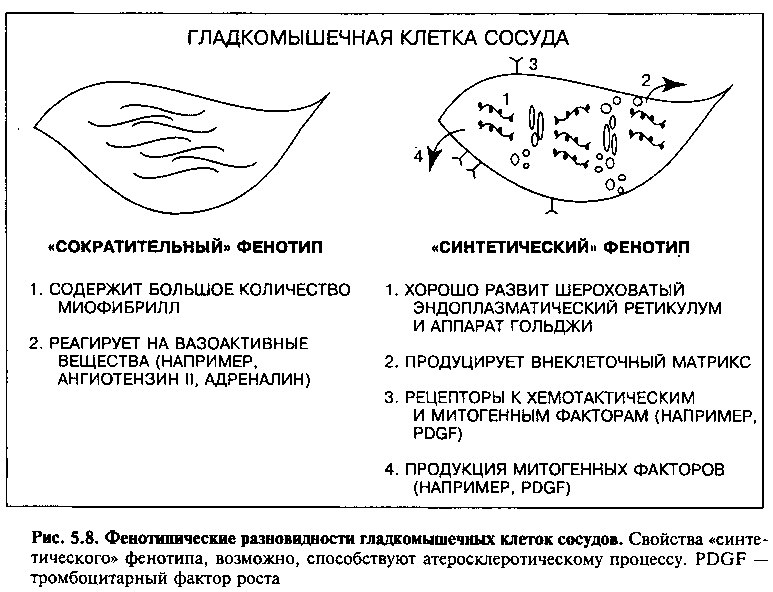
лин. Когда вазоактивный лиганд связывается со своим рецептором, миоцит сокращается или расслабляется, изменяя таким образом диаметр просвета сосуда и, соответственно, кровоток.
Напротив, гладкомышечные клетки «синтетического» фенотипа характеризуются меньшим количеством миофибрилл в цитоплазме. Зато в них хорошо представлен шероховатый эндоплазматический ретикулум и аппарат Гольджи, что отражает активный синтез белка. Гладкомышечные клетки этого фенотипа продуцируют коллаген, эластин и протеогликаны, которые формируют матрикс медии сосуда. Они также экспрессируют рецепторы для таких хемотактических и митогенных факторов как ТФР, в ответ на которые гладкомышечные клетки мигрируют из интимы в ме-дию и пролиферируют. «Синтетические» гладкомышечные клетки также могут продуцировать свои собственные митогенные факторы (включая ТФР), что, возможно, ведет к аутостимуляции и пролиферации.
Имеются данные, что гладкомышечные клетки внутри атеросклеротических поражений изменяют свой фенотип с сократительного на синтетический. Возможно, это изменение клеточной функции во многом определяет реакцию на воздействие ключевых агонистов, формирование экстрацеллюлярного матрикса и пролиферацию гладкомышечных клеток, характерную для формирующихся атеросклеротических бляшек.
ПАТОГЕНЕЗ АТЕРОСКЛЕРОЗА
Хотя весь путь развития атеросклеротических поражений остается до конца не понятым, некоторые ключевые ступени его идентифицированы (рис. 5.9). Атерогенез требует участия клеток сосудистой стенки (эндотелиальных и гладкомышечных клеток), циркулирующих форменных элементов крови (моноциты, тромбоциты) и определенных химических медиаторов (называемых цитокинами).
Дисфункция эндотелия
По мнению многих исследователей, событием, начинающим процесс атерогенеза, является «повреждение» эндотелия артерии. Существуют различные доказательства этого утверждения. Так, известно, что далеко зашедшие атеросклеротические поражения практически не встречаются на протяжении кровеносного сосуда, но часты в местах ветвления артерии. Это — зоны турбулентности, где существует наибольшая вероятность травмы эндотелия. Другое доказательство вытекает из перечисленных выше факторов риска атеросклероза. Общим механизмом действия для всех факторов является нарушение функционирования эндотелиальных клеток. Например, курение повышает уровень циркулирующего монооксида углерода и усугубляет тканевую гипоксию, что может повреждать эндотелий. Высокий уровень ЛНП или низкий ЛВП создают избыток холестерина, доступного для захвата интимой с возможным ее повреждением. Артериальная гипертензия непосредственно увеличивает гемодинамический стресс, испытываемый эндотелием. Третья группа доказательств первичности повреждения эндотелия в процессе атерогенеза связана с опытами на животных, в которых было показано развитие атером в ответ на повреждение эндотелия.
До недавнего времени считалось, что повреждение эндотелия, запускающее атеросклероз, требует физической травмы эндотелиальных клеток и потери их части. Сейчас известно, что наличие структурного повреждения эндотелия на самом деле не обязательно. Фактически ранние стадии развития атеросклеротического поражения, включая аккумуляцию липидов внутри артериальной стенки и миграцию моноцитов, проходят при интактной поверхности эндотелиальных клеток. Как мы увидим далее, эти ранние сдвиги обусловлены специфическими биохимическими изменениями и клеточными сигнальными путями, а не структурными изменениями эндотелиального слоя.
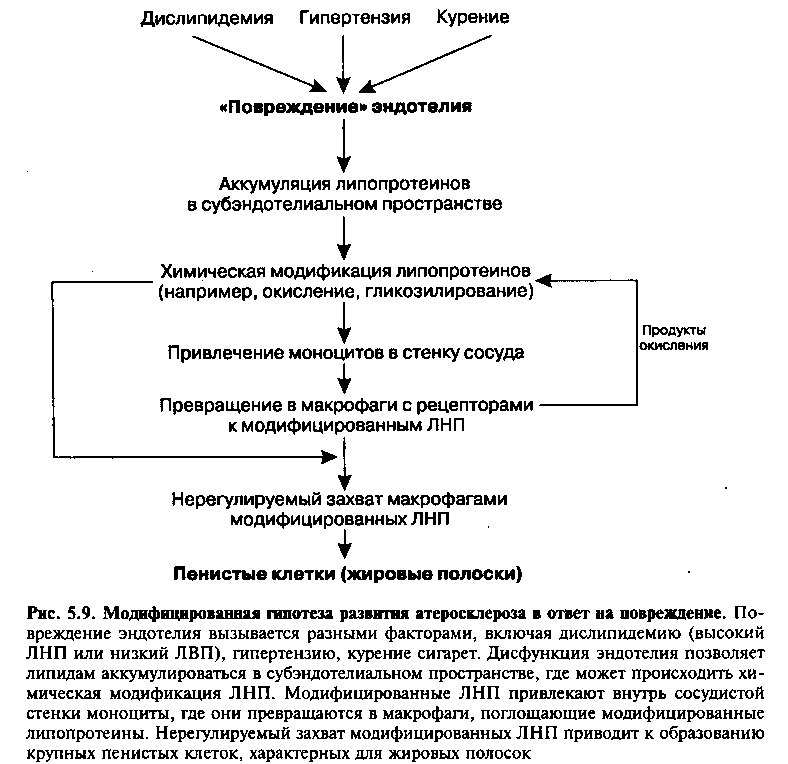
Многие факторы риска, ассоциированные с атеросклерозом (высокий холестерин ЛНП, курение, гипертензия, диабет) предрасполагают к дисфункции эндотелия. Даже до появления видимых атеросклеротических поражений эта дисфункция может проявляться: 1) нарушением барьерной функции эндотелия, 2) изменением его нормальной антитромботиче-ской активности, 3) нарушением высвобождения вазоактивных субстанций (например, простациклина и ЭЗФР-NO), что влияет на тонус подлежащих гладкомышечных клеток. Эти нежелательные последствия дисфункции эндотелия лежат в основе последующих событий атерогенеза.
Недавно в некоторых атеросклеротических поражениях были обнаружены определенные инфекционные агенты (например, из группы герпес-вирусов, хламидии), что вызвало вопрос об их возможной роли в развитии атерогенеза. Хотя эта взаимосвязь не доказана, некоторые исследователи полагают, что эти инфекционные агенты являются дополнительным источником повреждения эндотелия.
Гипотеза, согласно которой повреждение эндотелия и его дисфункция считаются инициирующим событием атерогенеза, известна как модифицированная гипотеза ответа на повреждение. Последовательность событий, которые следуют за.повреждением эндотелия, остается предметом продолжающегося изучения, и это изложение (схематически представленное на рис. 5.9), должно рассматриваться как общая схема, отдельные детали которой, скорее всего, при появлении новых знаний будут меняться.
Проникновение и модификация липопротеинов
При развитии дисфункции эндотелий более не может служить эффективным барьером на пути различных циркулирующих субстанций внутрь сосудистой стенки. Например, возросшая проницаемость эндотелия делает возможным транспорт липопротеинов низкой плотности внутрь интимы; этот процесс облегчается при повышении уровня циркулирующих ЛНП. Оказавшись внутри интимы, ЛНП скапливаются в субэндотелиальном пространстве, связываясь с компонентами внеклеточного матрикса. Попадание в эту «ловушку» удлиняет время нахождения ЛНП внутри сосудистой стенки, где липопротеин может подвергаться химическим модификациям, которые, видимо, являются решающим моментом в развитии атеросклеротических поражений.
Основной вид модификации ЛНП, происходящий в субэндотелиальном пространстве — это их окисление под действием присутствующих там свободных радикалов и ферментов. Источником прооксидантов являются, судя по всему, располагающиеся выше эндотелиальные клетки, а позднее — проникающие внутрь сосудистой стенки макрофаги. У больных сахарным диабетом с неконтролируемой гипергликемией другим важным путем биохимической модификации липопротеинов является их гликозилирование.
Биохимическая модификация ЛНП имеет два важных последствия: 1) модифицированные ЛНП (мЛНП) действуют как хемоаттрактанты, привлекающие циркулирующие моноциты внутрь сосудистой стенки, и 2) в отличие от нормальных ЛНП, мЛНП могут поглощаться макрофагами и другими клетками в больших количествах. Вспомните, что нормальный захват органами циркулирующих ЛНП регулируется рецепторами ЛНП и подчиняется механизму отрицательной обратной связи. Например, когда содержание холестерина внутри гепатоцита растет, число рецепторов ЛНП на поверхности клетки уменьшается, что ведет к уменьшению дальнейшего захвата липопротеина. Напротив, биохимически модифицированный ЛНП (окисленный или гликозилированный) не распознается рецептором ЛНП и не может попадать внутрь гепатоцита с помощью этого механизма. Однако мЛНП распознаются и захватываются с помощью рецепторов — макрофагов-«мусорщиков» и других клеток, и этот путь захвата не регулируется отрицательной обратной связью. Поэтому очень большие количества модифицированных ЛНП могут неконтролируемо поглощаться этими клетками, что приводит к появлению крупных «пенистых клеток» в жировых полосках.
Роль лейкоцитов
Вслед за проникновением и биохимической модификацией ЛНП следует другая ключевая ступень атерогенеза — привлечение лейкоцитов, в первую очередь моноцитов и Т-лимфоцитов, к сосудистой стенке. Факторами, участвующими в этом процессе, являются: 1) хемоатграктивные свойства мЛНП, 2) экспрессия специфических цитокинов (например, фактора некроза опухоли (ФНО) и хемоатграктивного белка моноцитов (ХАБМ), секретируемых поврежденными эндотелиальными клетками) и 3) экспрессия молекул адгезии на обращенной в просвет сосуда поверхности поврежденных эндотелиальных клеток. Примерами молекул адгезии являются молекула адгезии сосудистых клеток (VCAM-1) и межклеточная молекула адгезии (ICAM-1), представители суперсемейства генов иммуноглобулинов, и Р-селектин, особый вид молекул лейкоцитарных рецепторов. Показано, что компоненты окисленных ЛНП способствуют экспрессии генов VCAM, механически связывая накопление липидов в интиме артерии с появлением лейкоцитов. Гипотетическая роль молекул адгезии в привлечении моноцитов была подтверждена на животных. Например, атерогенная диета у кроликов приводит к экспрессии VCAM, предшествующей адгезии моноцитов к стенке сосуда.
После прилипания к поверхности интимы моноциты могут через межклеточные соединения эндотелиального монослоя проникнуть в субэндотелиальное пространство. Оказавшись под эндотелием, моноциты дифференцируются в макрофаги, фагоцитирующие клетки, способные поглощать модифицированные ЛНП в больших количествах (механизм участия клеток-«мусорщиков» см. выше). В результате макрофаги превращаются в нагруженные липидами пенистые клетки — первичный компонент жировой полоски.
Т-лимфоциты, основные медиаторы клеточного.иммунитета, также, видимо, играют важную роль на ранних стадиях атеросклероза. Т-клетки представлены относительно небольшой фракцией клеток внутри атероматозной бляшки, уступающей по численности макрофагам и гладкомышечным клеткам. Однако Т-клетки в процессе атерогенеза активируются и продуцируют цитокины, которые могут влиять на образование бляшки.
Роль гладкомышечных клеток
Трансформация жировой полоски в фиброзную бляшку включает в себя миграцию гладкомышечных клеток из медии в поврежденную интиму и секрецию ими большого количества соединительной ткани. Вещества, ответственные за миграцию и пролиферацию гладкомышечных клеток, в основном вырабатываются пенистыми клетками, активированными тромбоцитами и клетками эндотелия (рис. 5.10).
Пенистые клетки образуют несколько факторов, которые способствуют привлечению гладкомышечных клеток. Например, они выделяют тромбоцитарный фактор роста (ТФР), который стимулирует миграцию гладкомышечных клеток в субэндотелиальное пространство интимы с их
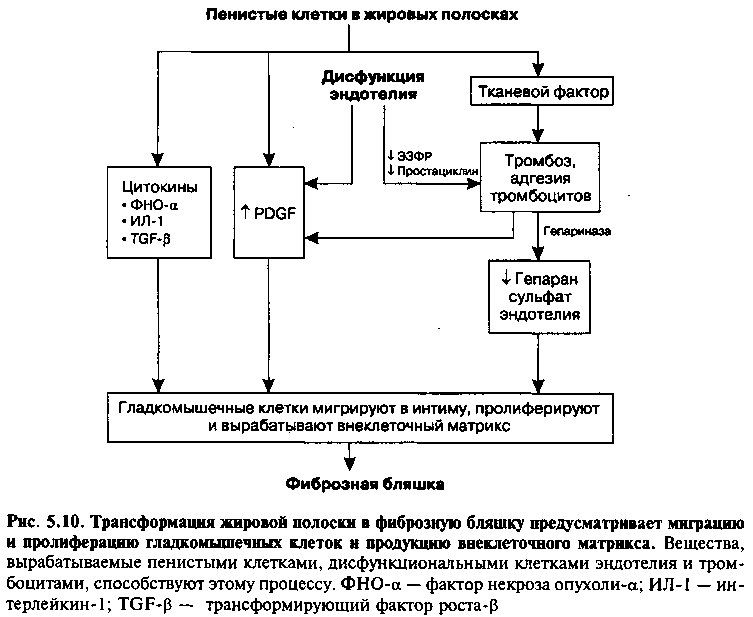
последующей репликацией там. Пенистые клетки также высвобождают цитокины и факторы роста (например, фактор некроза опухоли-а, интерлейкин-1, фактор роста фибробластов и трансформирующий фактор роста-p), которые дополнительно стимулируют пролиферацию гладкомышечных клеток и продукцию белков внеклеточного матрикса. Рост массы гладкомышечных клеток, прорастающих из медии в пораженную интиму, и развитие вокруг фиброзной покрышки из эктсрацеллюлярного матрикса характеризуют фиброзную бляшку — далеко зашедшую стадию атеросклероза.
Пенистые клетки продуцируют также большие количества тканевого фактора — протромбогенного вещества, которое при контакте с циркулирующей кровью активирует систему гемокоагуляции. Пока атероматозная бляшка покрыта фиброзной покрышкой и эндотелием, тромбогенное ядро, содержащее тканевой фактор, обычно «скрыто» от циркулирующей крови. Однако небольшие нарушения целостности далеко зашедших атеросклеротических поражений могут «раскрыть» тканевой фактор, вследствие чего богатые тромбоцитами микротромбы могут прилипать к поврежденной поверхности эндотелия. Активированные тромбоциты внутри таких микротромбов выделяют мощные факторы, которые также способствуют миграции и пролиферации гладкомышечных клеток. Эти факторы включают ТФР (чьи эффекты были описаны выше) и гепариназу. Последняя разрушает гепаран сульфат — полисахарид экстрацеллюлярного матрикса — который в норме ингибирует миграцию и пролиферацию гладкомышечных клеток.
Со своей стороны, поврежденная эндотелиальная клетка также играет роль в регуляции активности гладкомышечных клеток в развивающейся атероме. Поврежденные эндотелиальные клетки могут секретировать повышенные количества ТФР и пониженные — ЭЗФР-NO и простациклина, что также способствует привлечению и пролиферации гладкомышечных клеток.
Фиброзные бляшки нередко содержат центральное ядро из некротизированной ткани. Гибель клеток, возможно, происходит из-за токсических эффектов высокоокисленных ЛНП и накопления свободных радикалов. Ядро бляшки часто содержит дегенерирующие пенистые клетки, из которых высвобождаются кристаллы холестерина, которые потом могут окружаться врастающими в бляшку гладкомышечными клетками.
Таким образом, в настоящее время атеросклероз рассматривается как хронический воспалительный процесс в артериальной стенке, развивающийся в ответ на триггерные факторы, приводящие к дисфункции эндотелия, аккумуляции липидов внутри интимы, привлечению лейкоцитов и гладкомышечных клеток к интиме сосуда и отложению внеклеточного матрикса.
Следует отметить, что кроме модифицированной гипотезы «ответа на повреждение» были выдвинуты другие теории атерогенеза, включая и такие, в которых повреждение эндотелия рассматривается не как первопричина, а как результат. Например, «моноклональная гипотеза» предполагает, что первичным событием атерогенеза является миграция и пролиферация гладкомышечных клеток в интиму, индуцированная генетическими, химическими или вирусными стимулами. Этот взгляд поддерживается тем, что содержащиеся в некоторых атеросклеротических бляшках человека гладкомышечные клетки действительно происходят от одной клетки.
ОСЛОЖНЕНИЯ АТЕРОСКЛЕРОЗА
Атеросклеротические бляшки обычно растут постепенно и привлекают к себе внимание только при существенном уменьшении кровоснабжения какого-то органа или клинически значимом нарушении структуры стенки артерии. Основные клинические проявления атеросклероза связаны с ©сложенными фиброзными бляшками, как показано на рис. 5.2 и табл. 5.3. Например, как будет рассказано в следующей главе, атеросклеротические поражения коронарного русла могут нарушать перфузию миокарда и вызывать приступы дискомфорта в грудной клетке (стенокардию), классический симптом ИБС. В других случаях фиброзная бляшка может осложняться тромбозом, полностью перекрывающим просвет коронарной артерии, что приводит к острому инфаркту миокарда (см. главу 7).
Таблица 5,3, Осложнения атеросклероза
Осложнение |
Механизм |
Примеры |
Сужение и кальцификация сосуда |
Прогрессирующий рост фиброзной бляшки Организация микротромбов внутри бляшки Кровоизлияние в бляшку |
Ишемия миокарда (см. гл. 6) Перемежающаяся хромота (см. гл. 15) |
Тромбообразова-ние с окклюзией просвета |
Разрыв или изъязвление бляшки Кровоизлияние в бляшку с ее разрывом |
Инфаркт миокарда или нестабильная стенокардия Инфаркт мозга (ишемический инсульт) |
Периферические эмболии |
Фрагментация и перемещение атероматозного материала из больших проксимальных сосудов в меньшие периферические |
Эмболический инсульт Почечная недостаточность вследствие эмболии атероматозным материалом |
Снижение прочности сосудистой стенки |
Давление на прилежащую медию вызывает атрофию мышечных клеток и потерю эластической ткани |
Аневризмы аорты (см. гл. 15) |
Недавние исследования показали, что степень сужения коронарных артерий (определяемая ангиографически) не коррелирует вполне с последующим развитием инфарктов миокарда в местах наибольшего стенозирования. Это подтверждает гипотезу о том, что первичным механизмом развития острых коронарных синдромов является разрыв бляшки с последующим тромбозом, а не окклюзия сосуда в результате постепенно прогрессирующего увеличения фиброзной бляшки. «Бляшки-виновники», приводящие к острому тромбозу (и соответствующим клиническим проявлениям), часто не видны при ангиографии.
Факторы, делающие такую бляшку более подверженной разрыву, включают относительно тонкую фиброзную покрышку, отделяющую пенистые клетки (содержащие тканевой фактор, мощный прокоагулянт) от
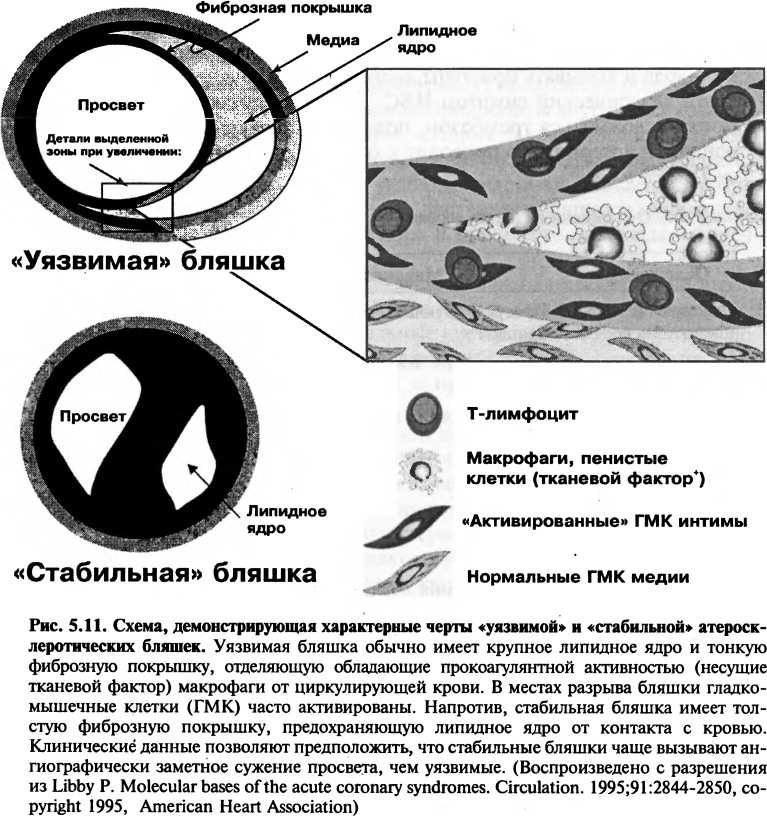
элементов циркулирующей крови (рис. 5.11). Эти «ранимые» бляшки часто имеют на макропрепарате маленькие размеры и в меньшей степени препятствуют кровотоку, чем хронические, стабильные бляшки с толстой фиброзной покрышкой, которые менее подвержены разрыву. «Уязвимые» места можно отличить по очень большому липидному ядру и высокой концентрации воспалительных клеток (макрофагов и Т-лимфоцитов). Макрофаги в подобных бляшках часто располагаются на границе бляшки и нормальной ткани (область «плеча») и секретируют медиаторы воспаления и ферменты, которые разрушают и ослабляют фиброзную покрышку, делая ее более подверженной разрыву. Кроме того, гамма-интерферон, медиатор, вырабатываемый Т-клетками в ответ на хроническое воспаление внутри бляшки, может ингибировать синтез коллагена гладкомышечными клетками, что нарушает их функцию поддержания целостности и репарации фиброзной покрышки, которая предохраняет бляшку от разрыва. Бляшки, способные синтезировать и сохранять толстую фиброзную капсулу, менее склонны к разрыву.
Как указывалось выше, недавние клинические исследования гиполи-пидемических препаратов продемонстрировали выраженное снижение частоты коронарных событий, не пропорциональное достигнутому регрессу стенозов коронарных артерий. Считается, что выраженное клиническое улучшение связано с уменьшением содержания липидов в атеросклеротической бляшке и, таким образом, ее «стабилизацией», что ведет к большей ее устойчивости к разрыву.
ЗАКЛЮЧЕНИЕ
В индустриализированном обществе атеросклероз вызывает больше смертей, чем любые другие заболевания. В процессе атерогенеза в интиме артерий мышечного типа накапливаются жировые отложения и фиброзная ткань. Наиболее ранним патоморфологическим проявлением атеросклероза являются жировые полосы. По-видимому, развитие атеросклеротических поражений обусловлено сложными взаимодействиями между клетками сосудистой стенки (эндотелиальными и гладкомышечными), циркулирующими клетками крови (лейкоцитами, тромбоцитами), липопротеинами и разными цитокинами и факторами роста. Клинические проявления атеросклероза развиваются в результате сужения просвета и кальцификации стенки сосуда, трещин или разрывов бляшки с последующим тромбозом, кровоизлияний в бляшку и ослабления стенки артерии.
Главные факторы риска развития атеросклероза включают дислипидемию (высокий ЛНП или низкий ЛВП), гипертензию, курение, раннее развитие коронарной болезни у родственников и диабет. Активное выявление и коррекция модифицируемых факторов риска является ключевым моментом современной профилактической кардиологии и скорее всего будет способствовать дальнейшему снижению сердечно-сосудистой заболеваемости и смертности.
Следующие две главы касаются двух наиболее важных осложнений атеросклероза: хронической ишемической болезни сердца и инфаркта миокарда.